Focus on Causality in ESC/iPSC-Based Modeling of Psychiatric Disorders

Abstract
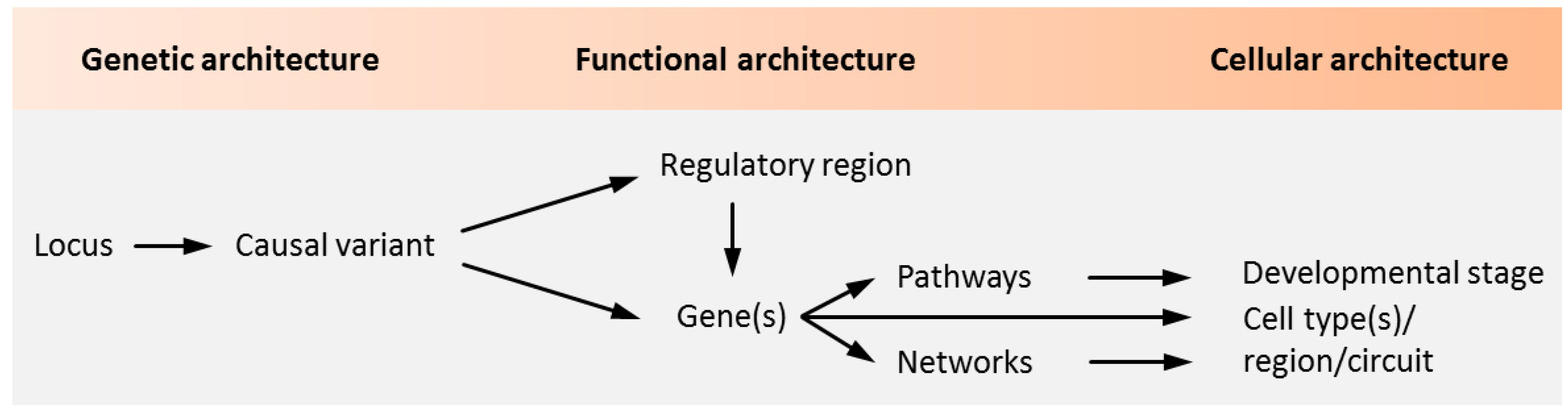
:1. Introduction
2. The Rationale of ESC/iPSC-Based Modeling of Psychiatric Disorders
3. ESC/iPSC-Based Modeling of Structural Variants
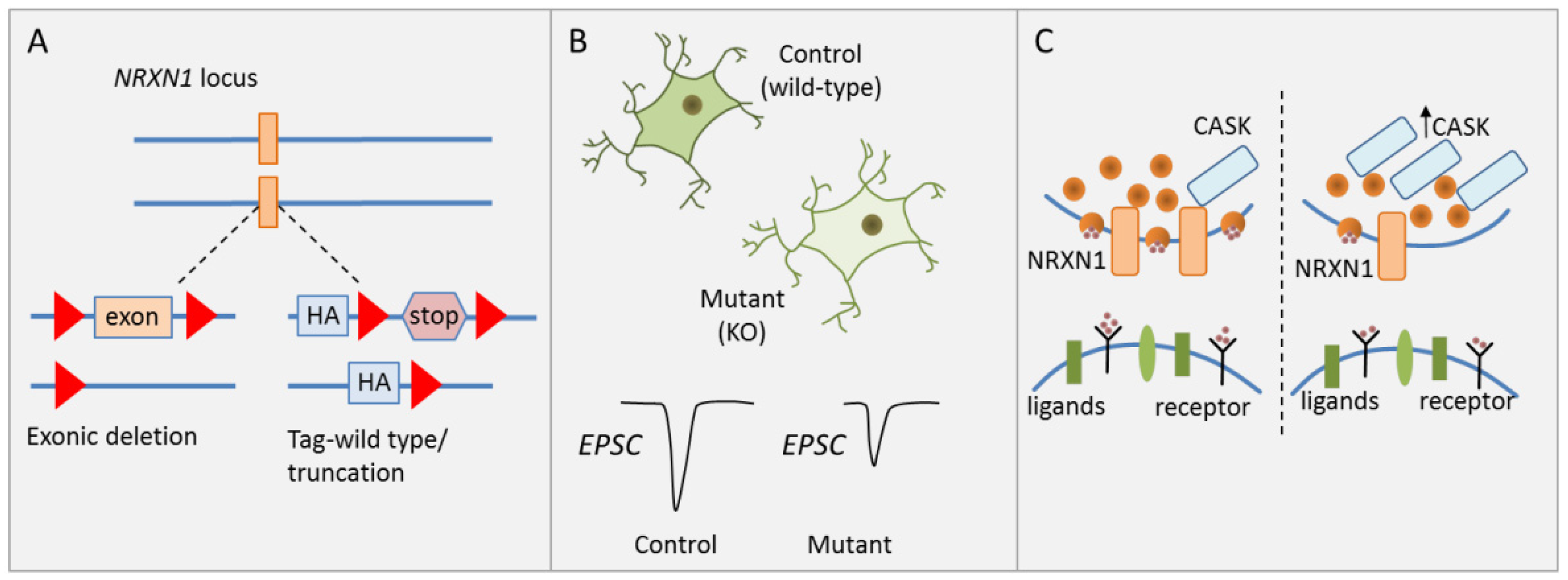
3.1. CNV Risk Variants at 2p16.3 Associate with Synaptic Transmission
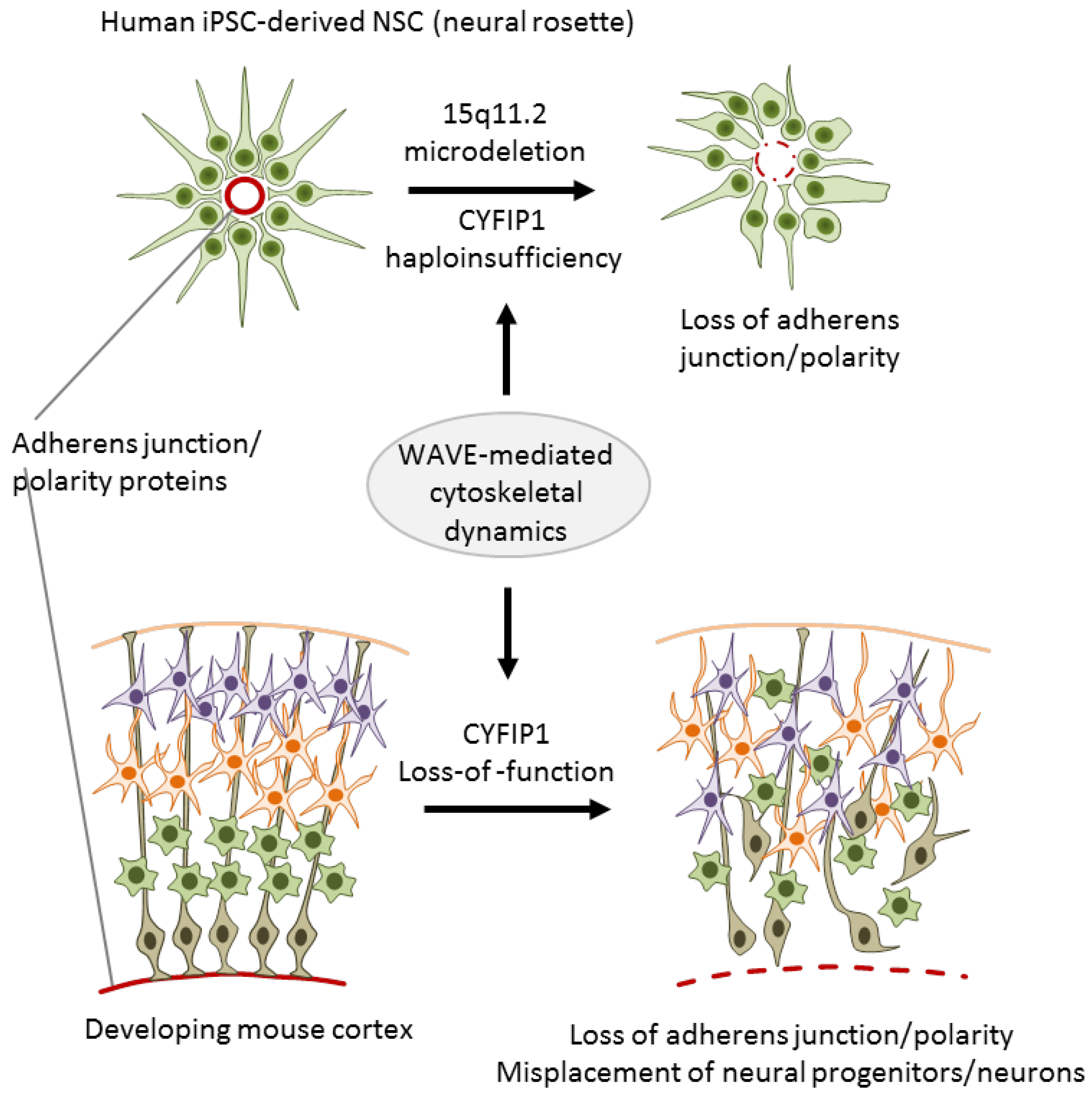
3.2. CNV Risk Variants at 15q11.2 Associate with NSC Adherens Junctions and Cell Polarity
3.3. Private CNVs at PCDH15 and RELN Impact Dendrite and Synapse Formation
3.4. DISC1′s Role in Presynaptic Function and Neural Development
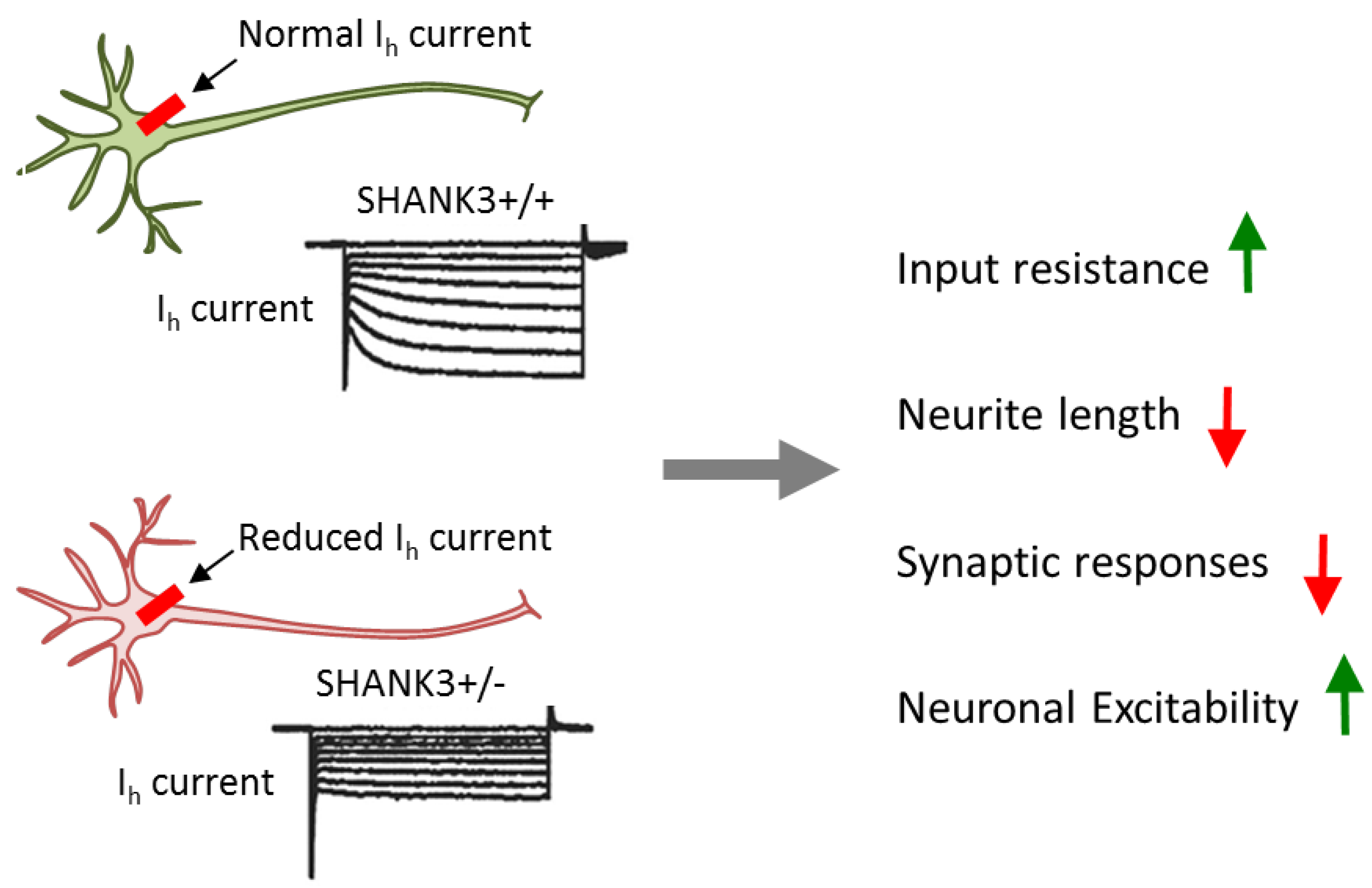
3.5. SHANK3 Mutations Impact Neuronal and Synaptic Functions
4. ESC/iPSC-Based Modeling of Common GWAS Risk Variants and Genes
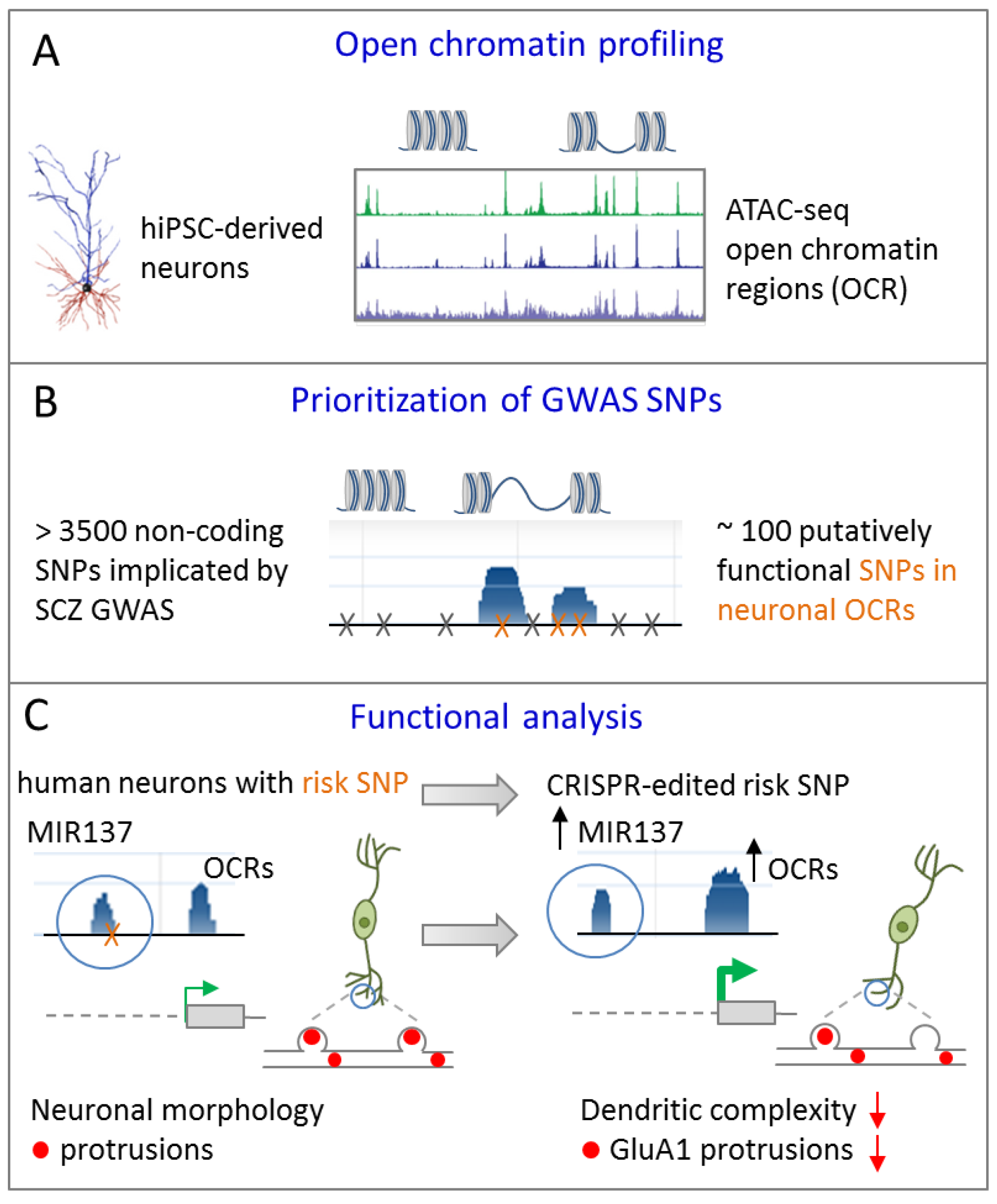
4.1. Credible SNP at miR-137 Regulates Dendritic Arborization
4.2. Joint Effects of Common GWAS Risk Variants on Gene Expression
4.3. Cataloging Credible Risk SNPs and Genes
5. Outlook and Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three dimensional |
| ADHD | Attention deficit hyperactivity disorder |
| ATAC-seq | Assay for transposase-accessible chromatin |
| ASD | Autism spectrum disorder |
| BD | Bipolar disorder |
| CASK | Calcium/calmodulin-dependent serine protein kinase |
| CLCN3 | Chloride voltage-gated channel 3 |
| CNV | Copy number variation |
| CNS | Central nervous system |
| CNTN4 | Contactin 4 |
| COS | Childhood onset of schizophrenia |
| CRISPR/CAS9 | Clustered regulatory interspaced short palindromic repeats/crispr-associated protein 9 |
| CRISPRa/i | CRISPR mediated gene activation or inhibition |
| CYFIP1 | Cytoplasmatic FMR1-interacting protein |
| DEG | Differentially expressed gene |
| DRD2 | Dopamine receptor D2 subtype |
| EB | Embryoid body |
| EPSC | Excitatory postsynaptic current |
| eQTL | Expression quantitative trait locus |
| ESC | Embryonic stem cell |
| FURIN | Paired basic amino acid cleaving enzyme |
| FOXG1 | Forkhead box G1 |
| FTO | a-Ketoglutarate-dependent dioxygenase |
| GABA | γ-aminobutyric |
| GWAS | Genome-wide association study |
| iPSC | Induced pluripotent stem cell |
| iN | Induced neuron |
| NPC | Neural progenitor cells |
| NRXN1 | Neurexin-1 |
| MDD | Major depressive disorder |
| MRPA | Massively parallel reporter assays |
| mEPSC | Miniature excitatory postsynaptic current |
| OCR | Open chromatin region |
| PCDH | Protocadherin |
| PIR | Promoter interacting region |
| PMDS | Phelan-McDermid syndrome |
| PSD | Postsynaptic density |
| RELN | Reelin |
| RGC | Radial glia cell |
| RNA-seq | RNA sequencing |
| SHANK | SH3 and ankyrin repeat domain |
| SNAP91 | Synaptosomal-associated protein 91-Kd |
| SNP | Single nucleotide polymorphism |
| SCZ | Schizophrenia |
| TF | Transcription factor |
| TSNARE1 | t-SNARE domain containing 1 |
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- United Nations, Department of Economic and Social Affairs Disability: Mental Health and Development. Available online: https://www.un.org/development/desa/disabilities/issues/mental-health-and-development.html (accessed on 18 September 2019).
- WHO Fact Sheets: Mental Health. Available online: http://www.who.int/topics/mental_health/factsheets/en/ (accessed on 18 September 2019).
- Stephan, K.E.; Bach, D.R.; Fletcher, P.C.; Flint, J.; Frank, M.J.; Friston, K.J.; Heinz, A.; Huys, Q.J.M.; Owen, M.J.; Binder, E.B.; et al. Charting the landscape of priority problems in psychiatry, part 1: Classification and diagnosis. Lancet Psychiatry 2016, 3, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.F.; Geschwind, D.H. Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 2019, 177, 162–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, A.M.; Sullivan, P.F.; Lewis, C.M. Uncovering the Genetic Architecture of Major Depression. Neuron 2019, 102, 91–103. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium Moller Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 2019, 179, 1469–1482.e11. [CrossRef] [Green Version]
- Girdhar, K.; Hoffman, G.E.; Jiang, Y.; Brown, L.; Kundakovic, M.; Hauberg, M.E.; Francoeur, N.J.; Wang, Y.-C.; Shah, H.; Kavanagh, D.H.; et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018, 21, 1126–1136. [Google Scholar] [CrossRef]
- O’Dushlaine, C. The Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 199–209. [Google Scholar]
- Bryois, J.; Skene, N.G.; Folkmann Hansen, T.; Kogelman, L.J.A.; Watson, H.J.; Brueggeman, L.; Breen, G.; Bulik, C.M.; Arenas, E.; Hjerling-Leffler, J.; et al. Genetic Identification of Cell Types Underlying Brain Complex Traits Yields Novel Insights Into the Etiology of Parkinson’s Disease. bioRxiv 2019, 528463. [Google Scholar] [CrossRef]
- Coleman, J.R.I.; Gaspar, H.A.; Bryois, J.; Breen, G. The genetics of the mood disorder spectrum: Genome-wide association analyses of over 185,000 cases and 439,000 controls. Biol. Psychiatry 2019, 19. [Google Scholar] [CrossRef]
- Collado-Torres, L.; Burke, E.E.; Peterson, A.; Shin, J.; Straub, R.E.; Rajpurohit, A.; Semick, S.A.; Ulrich, W.S.; BrainSeq Consortium; Price, A.J.; et al. Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron 2019, 103, 203–216. [Google Scholar] [CrossRef]
- Nagel, M.; Jansen, P.R.; Stringer, S.; Watanabe, K.; de Leeuw, C.A.; Bryois, J.; Savage, J.E.; Hammerschlag, A.R.; Skene, N.G.; Muñoz-Manchado, A.B.; et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018, 50, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polioudakis, D.; de la Torre-Ubieta, L.; Langerman, J.; Elkins, A.G.; Shi, X.; Stein, J.L.; Vuong, C.K.; Nichterwitz, S.; Gevorgian, M.; Opland, C.K.; et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 2019, 103, 785–801.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schork, A.J.; Won, H.; Appadurai, V.; Nudel, R.; Gandal, M.; Delaneau, O.; Revsbech Christiansen, M.; Hougaard, D.M.; Bækved-Hansen, M.; Bybjerg-Grauholm, J.; et al. A genome-wide association study of shared risk across psychiatric disorders implicates gene regulation during fetal neurodevelopment. Nat. Neurosci. 2019, 22, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Skene, N.G.; Bryois, J.; Bakken, T.E.; Breen, G.; Crowley, J.J.; Gaspar, H.A.; Giusti-Rodriguez, P.; Hodge, R.D.; Miller, J.A.; Muñoz-Manchado, A.B.; et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018, 50, 825–833. [Google Scholar] [CrossRef]
- Owen, M.J.; O’Donovan, M.C. Schizophrenia and the neurodevelopmental continuum: Evidence from genomics. World Psychiatry 2017, 16, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Florio, M.; Huttner, W.B. Neural progenitors, neurogenesis and the evolution of the neocortex. Development 2014, 141, 2182. [Google Scholar] [CrossRef] [Green Version]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2015, 20, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Burke, E.E.; Chenoweth, J.G.; Shin, J.H.; Collado-Torres, L.; Kim, S.K.; Micali, N.; Wang, Y.; Straub, R.E.; Hoeppner, D.J.; Chen, H.-Y. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, C.R.; Chen, J.; Tang, Y.; Southwell, D.G.; Chalmers, N.; Vogt, D.; Arnold, C.M.; Chen, Y.-J.J.; Stanley, E.G.; Elefanty, A.G.; et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 2013, 12, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.L.; de la Torre-Ubieta, L.; Tian, Y.; Parikshak, N.N.; Hernández, I.A.; Marchetto, M.C.; Baker, D.K.; Lu, D.; Hinman, C.R.; Lowe, J.K.; et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 2014, 83, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turksen, K.; Nagy, A. (Eds.) Induced Pluripotent Stem (iPS) Cells: Methods and Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; ISBN 978-1-4939-3055-5. [Google Scholar]
- Brennand, K.J.; Gage, F.H. Concise review: The promise of human induced pluripotent stem cell-based studies of schizophrenia. Stem Cells 2011, 29, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, L.; Rees, E.; Moran, J.L.; Chambert, K.D.; Milanova, V.; Craddock, N.; Purcell, S.; Sklar, P.; McCarroll, S.; Holmans, P.; et al. De novo CNVs in bipolar affective disorder and schizophrenia. Hum. Mol. Genet. 2014, 23, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Green, E.K.; Rees, E.; Walters, J.T.R.; Smith, K.-G.; Forty, L.; Grozeva, D.; Moran, J.L.; Sklar, P.; Ripke, S.; Chambert, K.D.; et al. Copy number variation in bipolar disorder. Mol. Psychiatry 2016, 21, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirov, G. CNVs in neuropsychiatric disorders. Hum. Mol. Genet. 2015, 24, R45–R49. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Howrigan, D.P.; Merico, D.; Thiruvahindrapuram, B.; Wu, W.; Greer, D.S.; Antaki, D.; Shetty, A.; Holmans, P.A.; Pinto, D.; et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 2017, 49, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 2015, 17, 316–328. [Google Scholar] [CrossRef] [Green Version]
- Yoon, K.-J.; Nguyen, H.N.; Ursini, G.; Zhang, F.; Kim, N.-S.; Wen, Z.; Makri, G.; Nauen, D.; Shin, J.H.; Park, Y.; et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 2014, 15, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Ishikawa, M.; Fujimori, K.; Maeda, T.; Kushima, I.; Arioka, Y.; Mori, D.; Nakatake, Y.; Yamagata, B.; Nio, S.; et al. In vitro modeling of the bipolar disorder and schizophrenia using patient-derived induced pluripotent stem cells with copy number variations of PCDH15 and RELN. eNeuro 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.-S.; Yoon, K.-J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ye, F.; Wen, Z.; Guo, Z.; Yu, C.; Huang, W.-K.; Rojas Ringeling, F.; Su, Y.; Zheng, W.; Zhou, G.; et al. Structural interaction between DISC1 and ATF4 underlying transcriptional and synaptic dysregulation in an iPSC model of mental disorders. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, P.; Han, K.; Callahan, D.G.; Makovkina, E.; Muratore, C.R.; Lalli, M.A.; Zhou, H.; Boyd, J.D.; Kosik, K.S.; Selkoe, D.J.; et al. Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep. 2015, 12, 1414–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, P.; Lagomarsino, V.N.; Pearse, R.V.; Liao, M.; Ghosh, S.; Nehme, R.; Seyfried, N.; Eggan, K.; Young-Pearse, T.L. Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl. Psychiatry 2018, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Danko, T.; Botelho, S.C.; Patzke, C.; Pak, C.; Wernig, M.; Südhof, T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016, 352, aaf2669. [Google Scholar] [CrossRef] [Green Version]
- Forrest, M.P.; Zhang, H.; Moy, W.; McGowan, H.; Leites, C.; Dionisio, L.E.; Xu, Z.; Shi, J.; Sanders, A.R.; Greenleaf, W.J.; et al. Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 2017, 21, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Schrode, N.; Ho, S.-M.; Yamamuro, K.; Dobbyn, A.; Huckins, L.; Matos, M.R.; Cheng, E.; Deans, P.J.M.; Flaherty, E.; Barretto, N.; et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet. 2019, 51, 1475–1485. [Google Scholar] [CrossRef]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; LaMarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef] [Green Version]
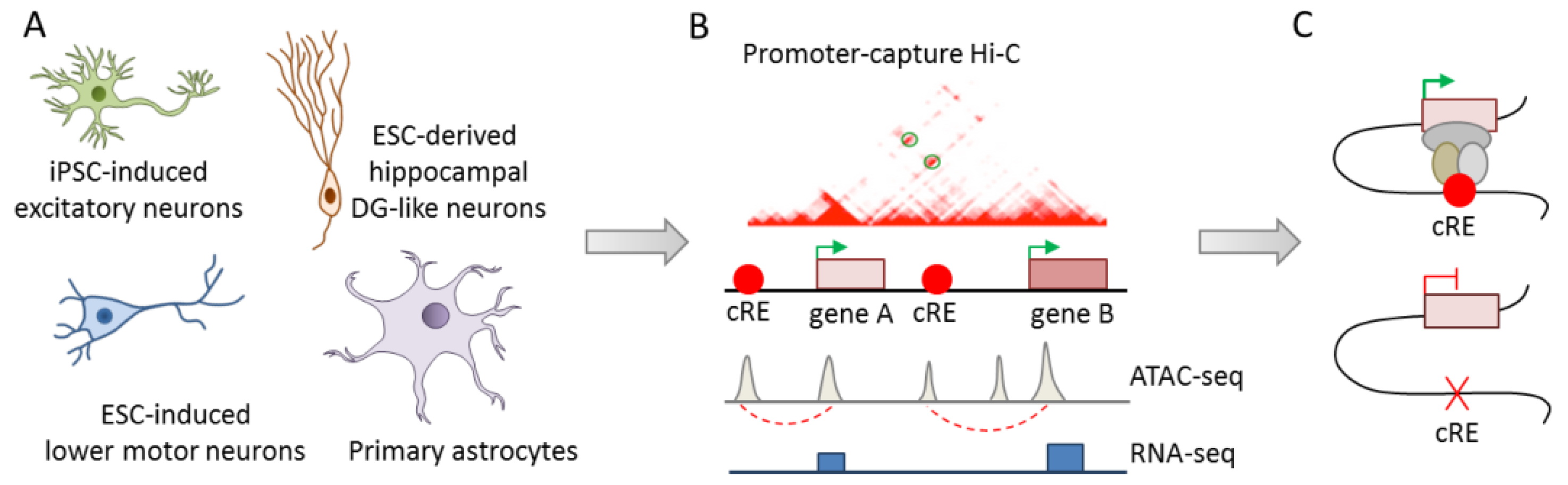
- Song, M.; Yang, X.; Ren, X.; Maliskova, L.; Li, B.; Jones, I.R.; Wang, C.; Jacob, F.; Wu, K.; Traglia, M.; et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019, 51, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, E.; Zhu, S.; Barretto, N.; Cheng, E.; Deans, P.J.M.; Fernando, M.B.; Schrode, N.; Francoeur, N.; Antoine, A.; Alganem, K.; et al. Neuronal impact of patient-specific aberrant NRXN1α splicing. Nat. Genet. 2019, 51, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Rees, E.; Walters, J.T.R.; Georgieva, L.; Isles, A.R.; Chambert, K.D.; Richards, A.L.; Mahoney-Davies, G.; Legge, S.E.; Moran, J.L.; McCarroll, S.A.; et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry 2014, 204, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Xiao, X.; Zhang, Z.; Li, M. Genetic insights and neurobiological implications from NRXN1 in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1400–1414. [Google Scholar] [CrossRef]
- Südhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Schrode, N.; Flaherty, E.; Brennand, K.J. New considerations for hiPSC-based models of neuropsychiatric disorders. Mol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Atasoy, D.; Schoch, S.; Ho, A.; Nadasy, K.A.; Liu, X.; Zhang, W.; Mukherjee, K.; Nosyreva, E.D.; Fernandez-Chacon, R.; Missler, M.; et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. USA 2007, 104, 2525–2530. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, H.; Meyer-Lindenberg, A.; Steinberg, S.; Magnusdottir, B.; Morgen, K.; Arnarsdottir, S.; Bjornsdottir, G.; Walters, G.B.; Jonsdottir, G.A.; Doyle, O.M.; et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014, 505, 361–366. [Google Scholar] [CrossRef] [Green Version]
- De Wolf, V.; Brison, N.; Devriendt, K.; Peeters, H. Genetic counseling for susceptibility loci and neurodevelopmental disorders: The del15q11.2 as an example. Am. J. Med. Genet. A 2013, 161A, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Lodato, S.; Shetty, A.S.; Arlotta, P. Cerebral cortex assembly: Generating and reprogramming projection neuron diversity. Trends Neurosci. 2015, 38, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noor, A.; Lionel, A.C.; Cohen-Woods, S.; Moghimi, N.; Rucker, J.; Fennell, A.; Thiruvahindrapuram, B.; Kaufman, L.; Degagne, B.; Wei, J.; et al. Copy number variant study of bipolar disorder in Canadian and UK populations implicates synaptic genes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-T.; Hinds, D.A.; Tung, J.Y.; Franz, C.; Fan, C.-C.; Wang, Y.; Smeland, O.B.; Schork, A.; Holland, D.; Kauppi, K.; et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat. Genet. 2017, 49, 152–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mountoufaris, G.; Canzio, D.; Nwakeze, C.L.; Chen, W.V.; Maniatis, T. Writing, Reading, and Translating the Clustered Protocadherin Cell Surface Recognition Code for Neural Circuit Assembly. Annu. Rev. Cell Dev. Biol. 2018, 34, 471–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costain, G.; Lionel, A.C.; Merico, D.; Forsythe, P.; Russell, K.; Lowther, C.; Yuen, T.; Husted, J.; Stavropoulos, D.J.; Speevak, M.; et al. Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum. Mol. Genet. 2013, 22, 4485–4501. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.; Kubo, K.-I.; Nakajima, K. Reelin and Neuropsychiatric Disorders. Front. Cell. Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Sobue, A.; Kushima, I.; Nagai, T.; Shan, W.; Kohno, T.; Aleksic, B.; Aoyama, Y.; Mori, D.; Arioka, Y.; Kawano, N.; et al. Genetic and animal model analyses reveal the pathogenic role of a novel deletion of RELN in schizophrenia. Sci Rep 2018, 8, 13046. [Google Scholar] [CrossRef]
- Shao, Z.; Noh, H.; Bin Kim, W.; Ni, P.; Nguyen, C.; Cote, S.E.; Noyes, E.; Zhao, J.; Parsons, T.; Park, J.M.; et al. Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat. Neurosci. 2019, 22, 229–242. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ziller, M.; Spengler, D. Progress in iPSC-Based Modeling of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 4896. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.F. Questions about DISC1 as a genetic risk factor for schizophrenia. Mol. Psychiatry 2013, 18, 1050–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-Y.; Liu, Y.; Yan, J.-W.; Hu, X.-L.; Zhu, D.-M.; Xu, X.-T.; Li, X.-S. Gene polymorphisms of DISC1 is associated with schizophrenia: Evidence from a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.A.; Sawa, A.; Holmes, S.E.; Ross, C.A.; DeLisi, L.E.; Margolis, R.L. A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol. Psychiatry 2005, 10, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Harony-Nicolas, H.; De Rubeis, S.; Kolevzon, A.; Buxbaum, J.D. Phelan McDermid Syndrome: From Genetic Discoveries to Animal Models and Treatment. J. Child Neurol. 2015, 30, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Champagne, N.; Lafrenière, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Côté, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Novella Romanelli, M.; Cerbai, E. The Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels: From Biophysics to Pharmacology of a Unique Family of Ion Channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Kathuria, A.; Nowosiad, P.; Jagasia, R.; Aigner, S.; Taylor, R.D.; Andreae, L.C.; Gatford, N.J.F.; Lucchesi, W.; Srivastava, D.P.; Price, J. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 2018, 23, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.X.; Crabtree, G.R.; Yoo, A.S. MicroRNAs: Regulators of neuronal fate. Curr. Opin. Cell Biol. 2013, 25, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Rajman, M.; Schratt, G. MicroRNAs in neural development: From master regulators to fine-tuners. Development 2017, 144, 2310–2322. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, R.M.; Gururajan, A.; Dinan, T.G.; Kenny, P.J.; Cryan, J.F. All Roads Lead to the miRNome: miRNAs Have a Central Role in the Molecular Pathophysiology of Psychiatric Disorders. Trends Pharmacol. Sci. 2016, 37, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; Absher, D.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs.Cross-Disorder Group of the Psychiatric Genomics Consortium. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [PubMed] [Green Version]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kähler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.; Wang, W.; Tsai, L.-H. Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol. Psychiatry 2013, 18, 11–12. [Google Scholar] [CrossRef]
- Lett, T.A.; Chakravarty, M.M.; Chakavarty, M.M.; Felsky, D.; Brandl, E.J.; Tiwari, A.K.; Gonçalves, V.F.; Rajji, T.K.; Daskalakis, Z.J.; Meltzer, H.Y.; et al. The genome-wide supported microRNA-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol. Psychiatry 2013, 18, 443–450. [Google Scholar] [CrossRef]
- Siegert, S.; Seo, J.; Kwon, E.J.; Rudenko, A.; Cho, S.; Wang, W.; Flood, Z.; Martorell, A.J.; Ericsson, M.; Mungenast, A.E.; et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 2015, 18, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421. [Google Scholar]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Ziller, M.; Spengler, D. The Future is The Past: Methylation QTLs in Schizophrenia. Genes (Basel) 2016, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Vockley, C.M.; Guo, C.; Majoros, W.H.; Nodzenski, M.; Scholtens, D.M.; Hayes, M.G.; Lowe, W.L.; Reddy, T.E. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 2015, 25, 1206–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulirsch, J.C.; Nandakumar, S.K.; Wang, L.; Giani, F.C.; Zhang, X.; Rogov, P.; Melnikov, A.; McDonel, P.; Do, R.; Mikkelsen, T.S. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 2016, 165, 1530–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, Y.; Zhang, Q.; Wu, J.; Liang, J.; Yu, S.; Wei, G.-H.; White, K.P.; Wang, X. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017, 18, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Xia, J.-H.; Zhu, J.; Gao, P.; Tian, Y.-J.; Du, M.; Guo, Y.-C.; Suleman, S.; Zhang, Q.; Kohli, M. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 2022. [Google Scholar] [CrossRef] [Green Version]
- van Arensbergen, J.; Pagie, L.; FitzPatrick, V.D.; de Haas, M.; Baltissen, M.P.; Comoglio, F.; van der Weide, R.H.; Teunissen, H.; Võsa, U.; Franke, L.; et al. High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 2019, 51, 1160–1169. [Google Scholar] [CrossRef]
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods Groups—Analysis Working Group; Enhancing GTEx (eGTEx) Groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481. [Google Scholar] [CrossRef]
- Mumbach, M.R.; Satpathy, A.T.; Boyle, E.A.; Dai, C.; Gowen, B.G.; Cho, S.W.; Nguyen, M.L.; Rubin, A.J.; Granja, J.M.; Kazane, K.R.; et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017, 49, 1602. [Google Scholar] [CrossRef] [Green Version]
- Smemo, S.; Tena, J.J.; Kim, K.-H.; Gamazon, E.R.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.-H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Torre-Ubieta, L.; Stein, J.L.; Won, H.; Opland, C.K.; Liang, D.; Lu, D.; Geschwind, D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 2018, 172, 289–304.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2018, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal Cortical Dendritic Spine Pathology in Schizophrenia and Bipolar Disorder. JAMA Psychiatry 2014, 71, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, J.E.; Goldstein, E.S.; Kilpatrick, S.T.; Lewin, B. Lewin’s genes XII; Jones & Bartlett Learning: Burlington, MA, USA, 2018; ISBN 978-1-284-10449-3. [Google Scholar]
- Bassetti, C.L.A.; Adamantidis, A.; Burdakov, D.; Han, F.; Gay, S.; Kallweit, U.; Khatami, R.; Koning, F.; Kornum, B.R.; Lammers, G.J.; et al. Narcolepsy-clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nat. Rev. Neurol. 2019, 15, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Fulco, C.P.; Nasser, J.; Jones, T.R.; Munson, G.; Bergman, D.T.; Subramanian, V.; Grossman, S.R.; Anyoha, R.; Doughty, B.R.; Patwardhan, T.A.; et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019, 51, 1664–1669. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Spengler, D. Genetic variation in the epigenetic machinery and mental health. Current Psychiatry Rep. 2012, 14, 138–149. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. Chromatin Remodeling Complex NuRD in Neurodevelopment and Neurodevelopmental Disorders. Front. Genet. 2019, 10, 682. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Sportelli, V.; Ziller, M.; Spengler, D.; Hoffmann, A. Tracing Early Neurodevelopment in Schizophrenia with Induced Pluripotent Stem Cells. Cells 2018, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Ziller, M.; Spengler, D. Childhood-Onset Schizophrenia: Insights from Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 3829. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish. Int. J. Mol. Sci. 2018, 19, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Mei, A.; Paquola, A.C.M.; Stern, S.; Bardy, C.; Klug, J.R.; Kim, S.; Neshat, N.; Kim, H.J.; Ku, M.; et al. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell 2018, 22, 684–697.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Paşca, S.P. Assembling human brain organoids. Science 2019, 363, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 2019, 25, 558–569.e7. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref | Variation | Gene | System | Cell Type | Key Findings |
|---|---|---|---|---|---|
| [31] | CNV | NRXN1 | Isogenic hESCS, conditional | Cortical glutamatergic | Impaired presynaptic neurotransmission, reduced spontaneous |
| (2p16.3) | heterozygous mutations | iNS, cortical neurons | mEPSC frequency and EPSC amplitude | ||
| [32] | CNV | CYFIP1 | Case/control-iPSCs | Neural rosette derived | Reduced adherens junctions and apical-basal polarity, gain- and |
| (15q11.3) | NPCs | loss-of-function experiments in vitro and in mice support a causal | |||
| role of CYFIP1 | |||||
| [33] | CNV | PCDH15 | Case-control iPSCs, isogenic | EBs, glutamatergic and | Structural changes in iPSC-derived neurons from patients are |
| RELN1 | homozygous mutations in | GABAergic iNS | recapitulated by isogenic iPSC-derived neurons, heightened | ||
| iPSCs from controls | AMPA and GABA receptor sensitivity in patients’ neurons | ||||
| [34] | 4-bp deletion | DISC1 | Case-control iPSCs, isogenic | EB-derived mixed | Impaired presynaptic function in neurons derived from patient |
| heterozygous mutation in | forebrain neurons | and gene-edited control iPSCs. Defect is rescued in neurons from | |||
| control iPSCs, isogenic | gene-edited patient iPSCs. DEGs are enriched in genes important | ||||
| rescue in patient iPSCs | to synapse and neurodevelopment and psychiatric disorders | ||||
| [35] | 4-bp deletion | DISC1 | Case/control-iPSCs | EB-derived mixed | Interaction between mutant DISC1 and ATF4 mediates aberrant |
| forebrain neurons | gene expression that is rescued by forced ATF4 expression. | ||||
| [36] | Exon 2 and | DISC1 | Isogenic homozygous exon 2 | EB-derived mixed | Mutations drive shift in dorsal fate owing to heightened Wnt- |
| exon 8 | or heterozygous exon 8 | forebrain neurons | signaling. This promotes premature NPC differentiation and | ||
| deletions | impaired cortical layer formation. Rescue by Wnt-antagonists | ||||
| [37] | [34,36] | DISC1 | See [34,36] | Cortical glutamatergic | Both mutations converge on few DEGs including UNCD5. |
| iNS | Reduced neurite outgrowth is rescued by UNCD5 activation and | ||||
| phenocopied by UNCD5 knockdown in wild-type neurons | |||||
| [38] | deletion | SHANK3 | Case/control-iPSC | Mixed forebrain | Increased input resistance and impaired excitatory transmission |
| neurons | associate with less excitatory synapses and glutamate receptors. | ||||
| SHANK3 complementation rescues only impaired transmission. | |||||
| [39] | deletion | SHANK3 | Isogenic hESCs, conditional | Cortical glutamatergic | Impaired synaptic transmission, dendritic arborization, and intrin- |
| heterozygous mutations | iNS | sic electrical properties owing to perturbed interaction | |||
| between SHANK3 and HCN channels in the postsynaptic density | |||||
| [40] | SNP | MIR137 | Isogenic iPSC from patient | Cortical glutamatergic | Credible SNP reduces miR-137 expression and leads to a more |
| with SCZ | iNS | mature neuronal phenotype in vitro compared to the gene-edited | |||
| non-risk allele | |||||
| [41] | SNP | FURIN, | Isogenic iPSCs from healthy | Cortical glutamatergic | Credible SNP in FURIN reduces neurite length and neuronal |
| TSNARE1 | donors | iNs (NPCs and mature | activity in excitatory iNs. CRISPRa/i regulation of multiple risk | ||
| CLCN3 | neurons), GABAergic | genes converges on synaptic abnormalities, shows synergistic | |||
| SNAP91 | iNs, induced astrocytes | effects, and recapitulates changes from SCZ postmortem brains | |||
| [42] | SNP | FOXG1 | Primary fetal progenitors | Primary fetal neurons | Credible SNP 760 kb away from FOXG1 regulates FOXG1 |
| expression via chromatin contact | |||||
| [43] | SNP | Isogenic iPSC from healthy | Cortical NPCs | Credible SNPs in GWAS loci regulate gene expression of ASCL1, | |
| donors | EFNB1, and MATR3 via long range chromatin interaction | ||||
| [44] | SNP | Isogenic iPSC from healthy | Excitatory and lower | PIRs are enriched for genes with a role in cell fate commitment | |
| donors | motor iNS, dendate | and for risk SNPs from major psychiatric disorders in a disease- | |||
| gyrus like neurons, | and cell type-specific manner. Monoallelic deletion of the PIR at | ||||
| primary fetal astrocytes | DRD2 causes downregulation in excitatory iNs | ||||
| [45] | CNV | NRXN1 | Case-control iPSCs | Glutamatergic (NGN2) | Heterozygous NRXN2 deletions cause quantitative and qualitative |
| (2p16.3) | and GABAergic | perturbations in isoform expression that associate with impaired | |||
| (ASCL2/DLX2) iNS | neuronal and synaptic function. De novo isoforms arising from | ||||
| NRXN1 deletions act potentially in a dominant negative mode. | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Ziller, M.; Spengler, D. Focus on Causality in ESC/iPSC-Based Modeling of Psychiatric Disorders. Cells 2020, 9, 366. https://doi.org/10.3390/cells9020366
Hoffmann A, Ziller M, Spengler D. Focus on Causality in ESC/iPSC-Based Modeling of Psychiatric Disorders. Cells. 2020; 9(2):366. https://doi.org/10.3390/cells9020366
Chicago/Turabian StyleHoffmann, Anke, Michael Ziller, and Dietmar Spengler. 2020. "Focus on Causality in ESC/iPSC-Based Modeling of Psychiatric Disorders" Cells 9, no. 2: 366. https://doi.org/10.3390/cells9020366