Human Skin Keratinocytes on Sustained TGF-β Stimulation Reveal Partial EMT Features and Weaken Growth Arrest Responses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
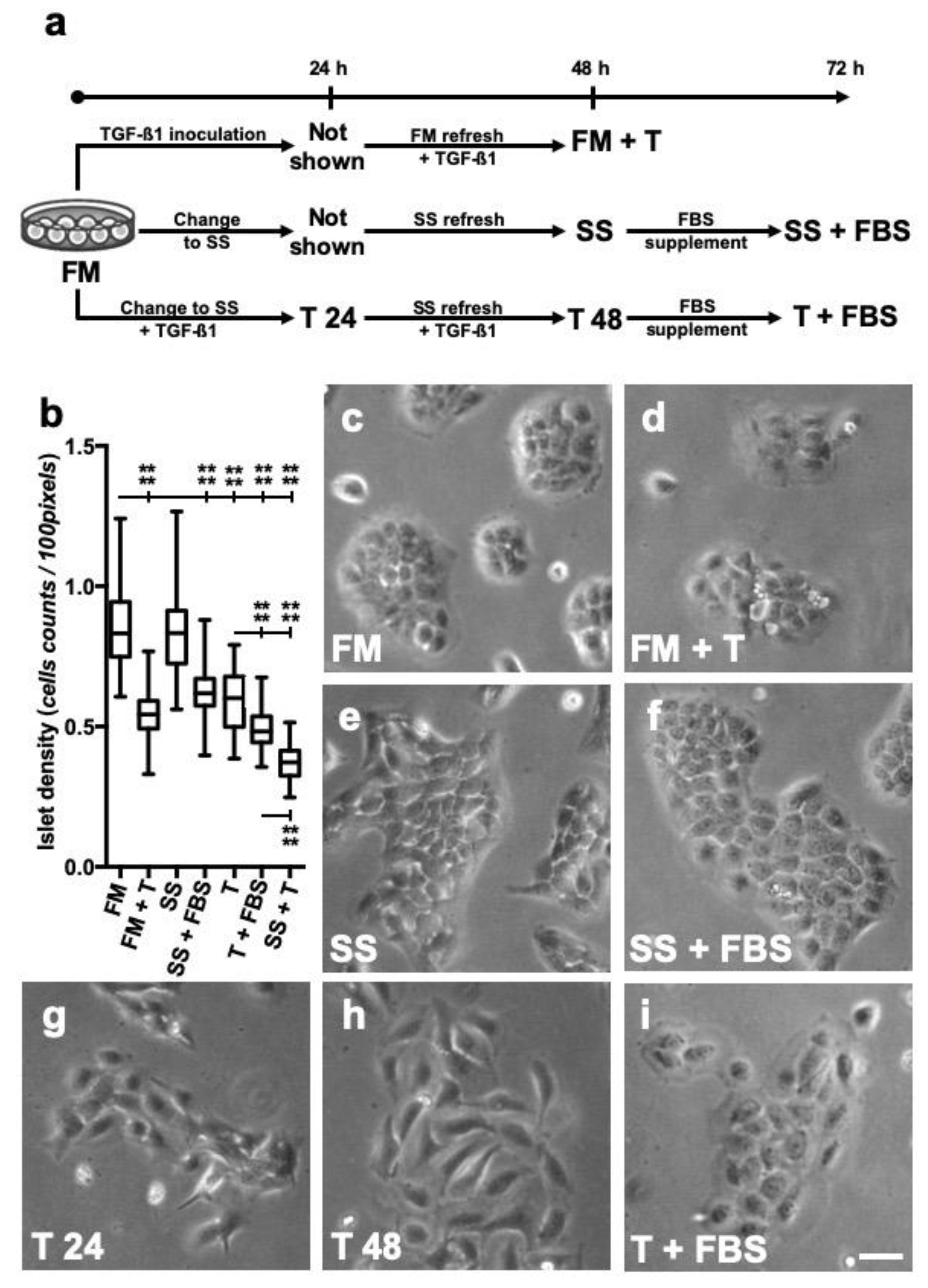
2.1. Cells Culture Conditions and Treatments
2.2. Cell Morphology and Islet Cell Density
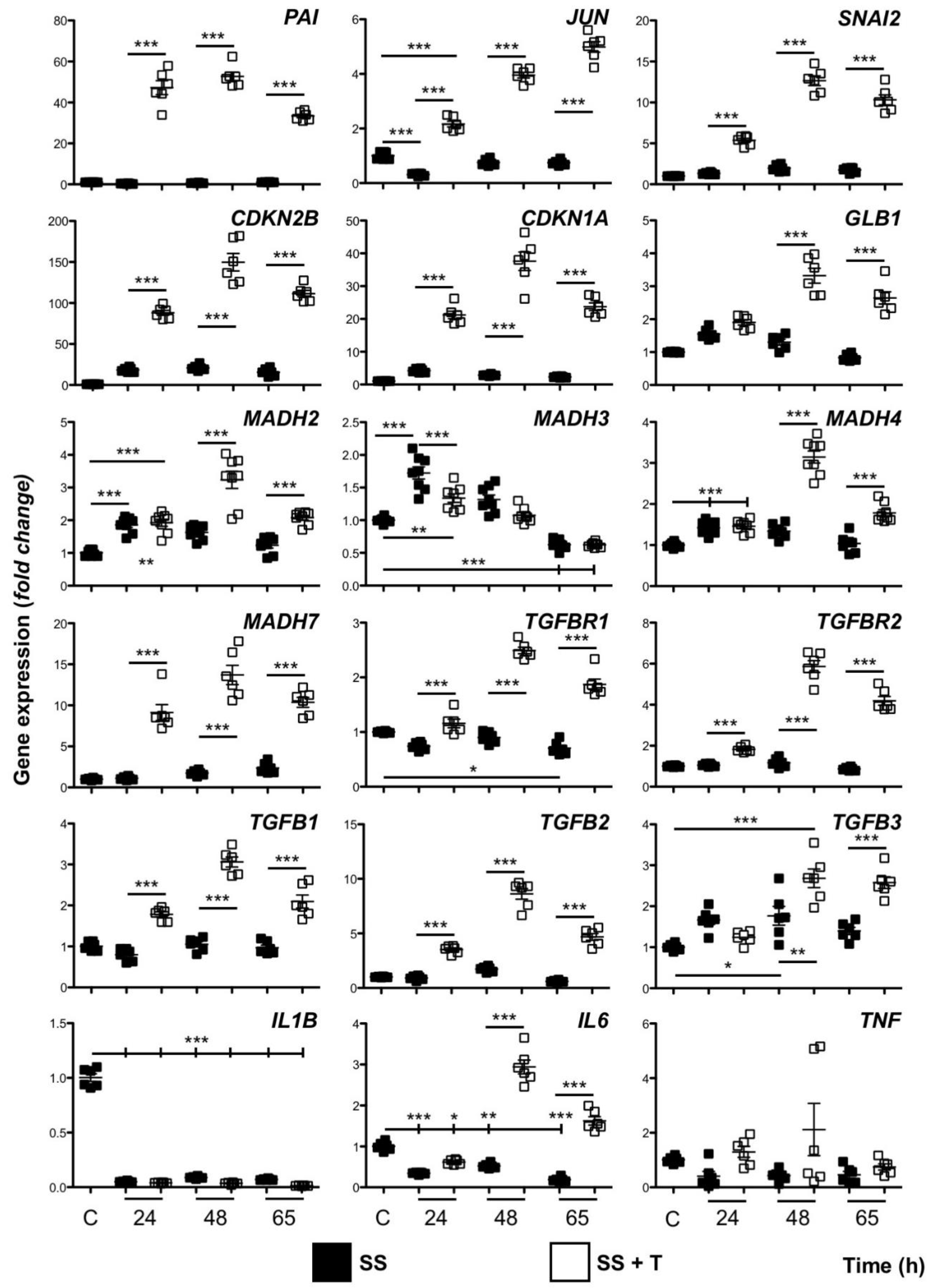
2.3. Quantitative PCR
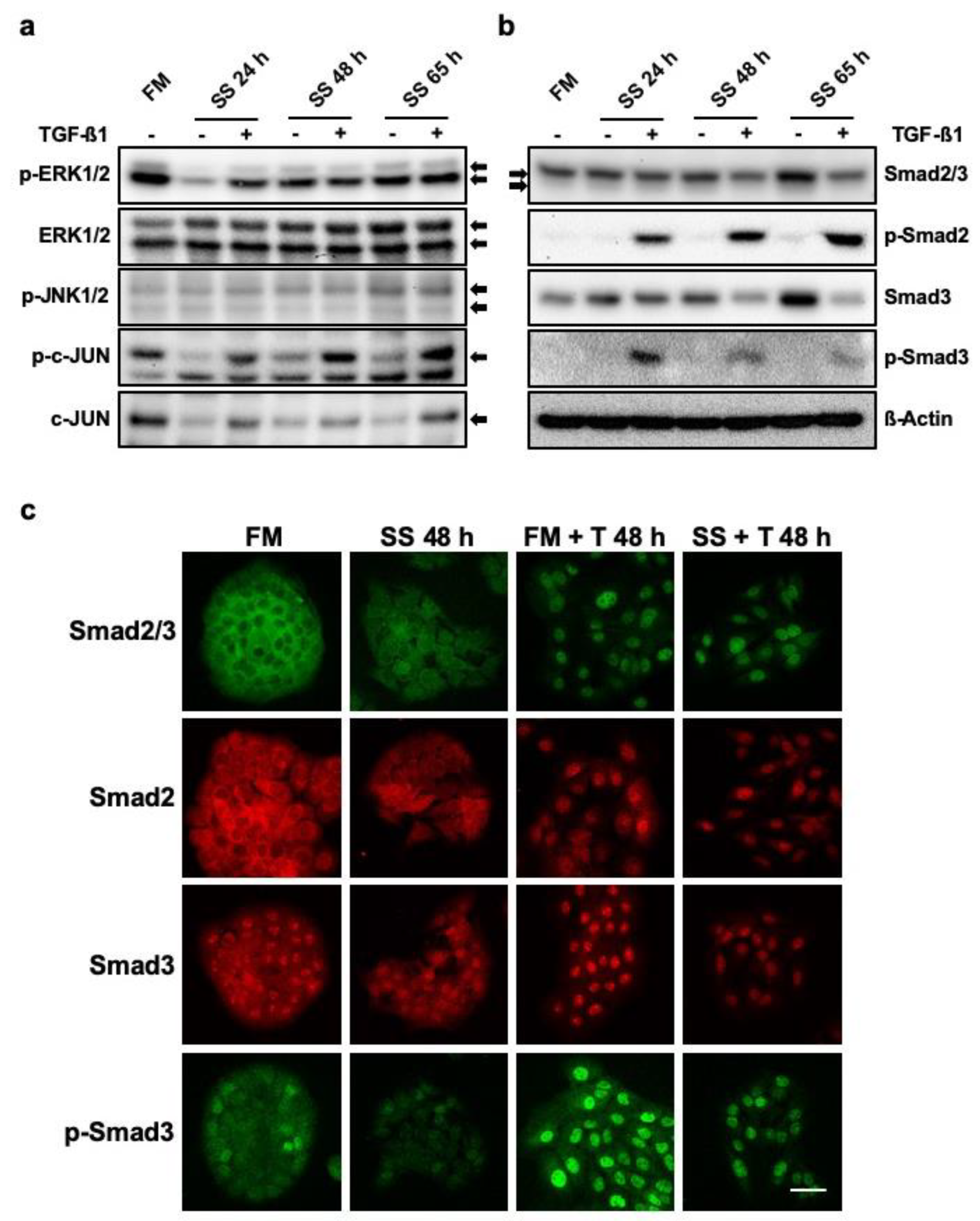
2.4. Western Blot
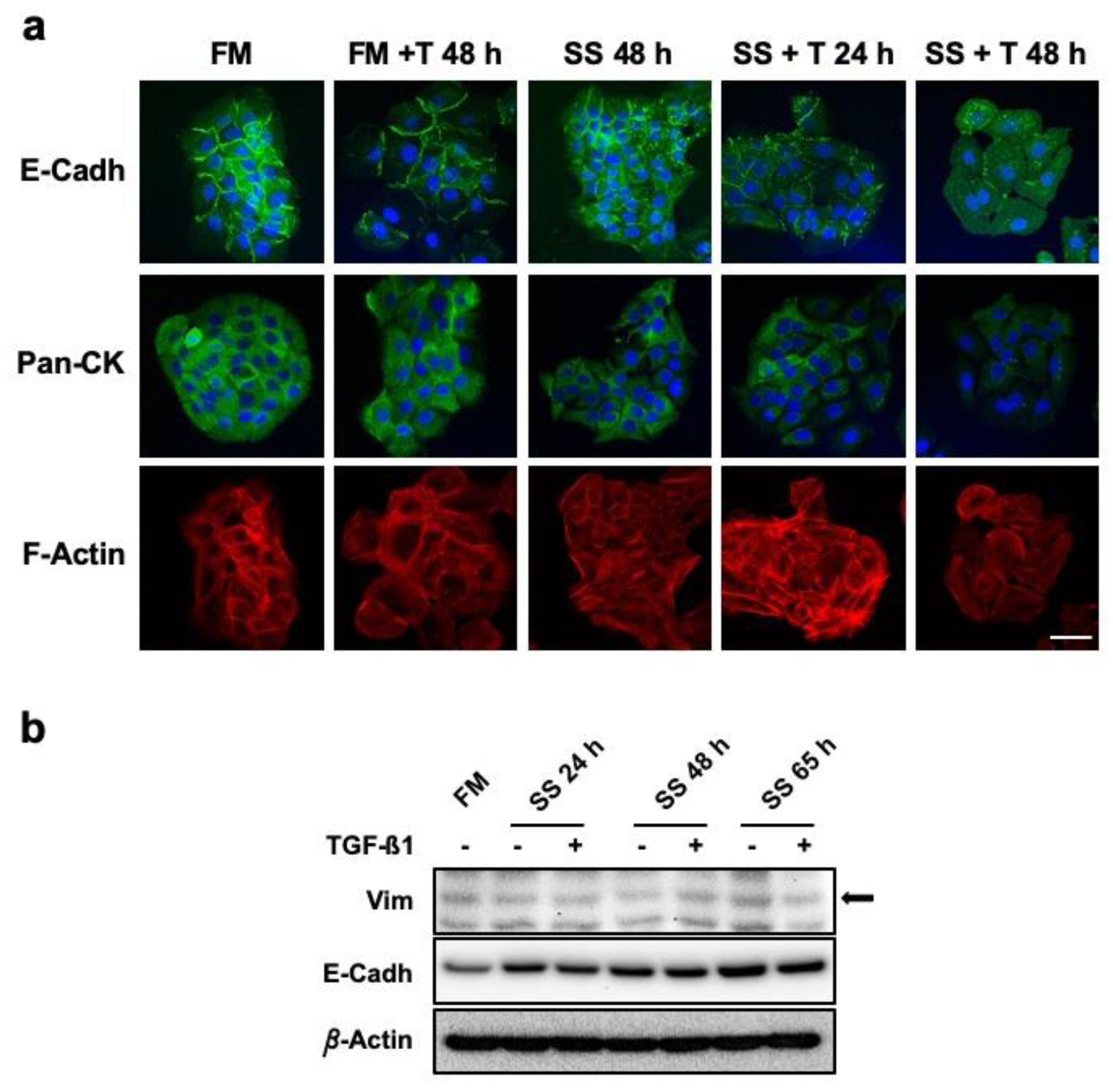
2.5. Immunocytochemistry and Image Analysis
2.6. Cell Cycle and Apoptosis Analysis
2.7. Statistics
3. Results
3.1. Long-Term TGF-β1 Exposure Alters the Conformation of HaCaT Cells
3.2. HaCaT Cells Continuously Exposed to TGF-β1 Exhibit a Distinct Gene Expression Profile
3.3. Serum-Deprived Hacat Cells Exposed in the Long Term to TGF-Β1 Do not Experience Complete Epithelial-to-Mesenchymal Transition
3.4. HaCaT Cells Long Term Cultured in SS And TGF-β1 Show Down-Regulation of Smad3 Protein Levels and Present with Altered Signalling
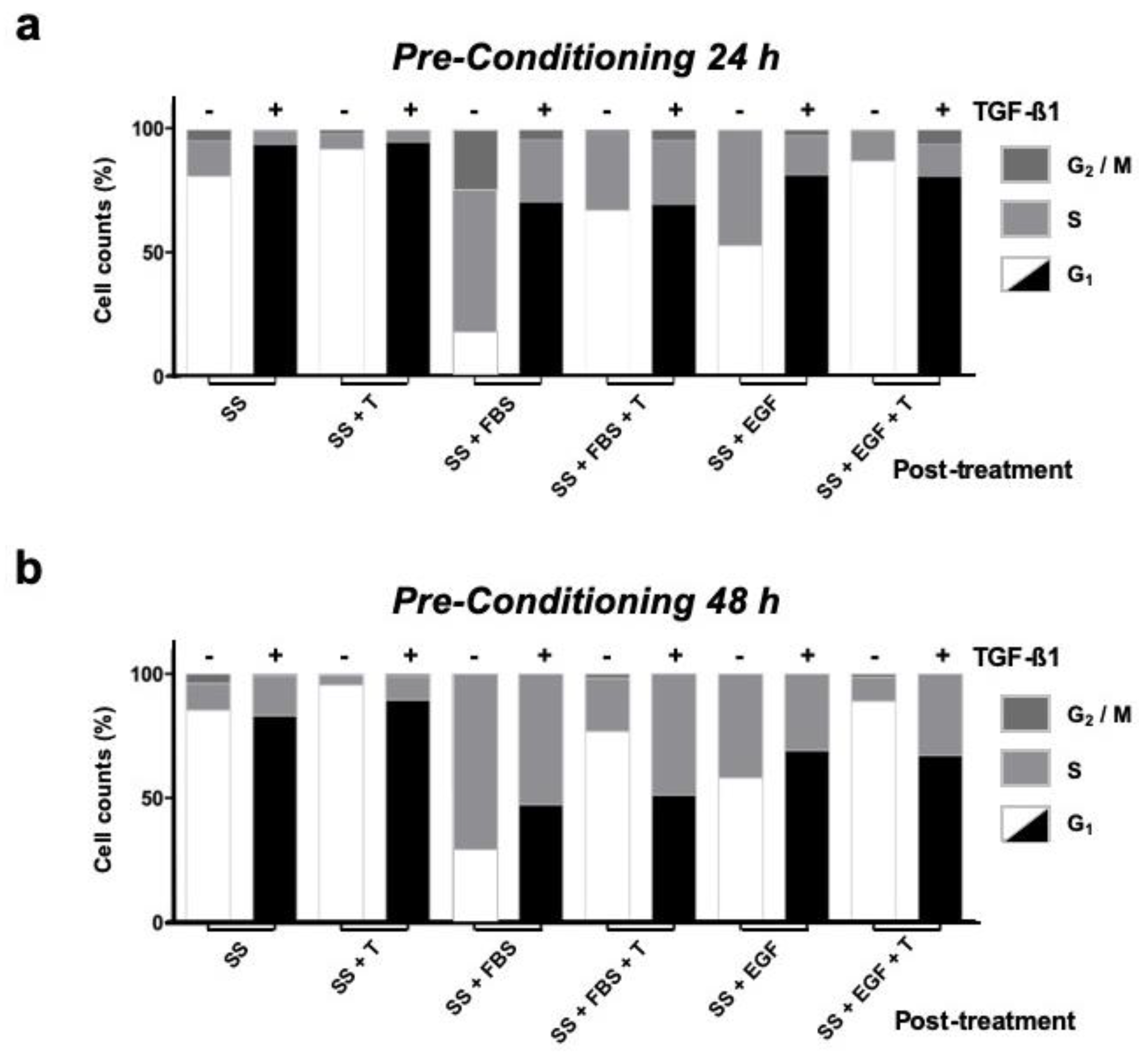
3.5. HaCaT Cells Exposed in the Long Term to TGF-β1 Show Altered Cell-Cycle Arrest Responses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Valluru, M.; Staton, C.A.; Reed, M.W.; Brown, N.J. Transforming Growth Factor-beta and Endoglin Signaling Orchestrate Wound Healing. Front. Physiol. 2011, 2, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.C.; Kim, H.T.; Park, S.H.; Cha, J.S.; Yufit, T.; Kim, S.J.; Falanga, V. Fibroblasts from chronic wounds show altered TGF-beta-signaling and decreased TGF-beta Type II receptor expression. J. Cell. Physiol. 2003, 195, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, S.; Ferguson, M.W. Transforming growth factor beta s and wound healing. Int. J. Biochem. Cell Biol. 1997, 29, 63–78. [Google Scholar] [CrossRef]
- Han, G.; Williams, C.A.; Salter, K.; Garl, P.J.; Li, A.G.; Wang, X.J. A role for TGFbeta signaling in the pathogenesis of psoriasis. J. Investig. Dermatol. 2010, 130, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 2007, 13, 3056–3062. [Google Scholar] [CrossRef]
- Finnson, K.W.; Arany, P.R.; Philip, A. Transforming Growth Factor Beta Signaling in Cutaneous Wound Healing: Lessons Learned from Animal Studies. Adv. Wound Care 2013, 2, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Schor, S.L.; Hinck, A.P. Biological activity differences between TGF-beta1 and TGF-beta3 correlate with differences in the rigidity and arrangement of their component monomers. Biochemistry 2014, 53, 5737–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Gilbert, R.W.D.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef]
- Finnson, K.W.; McLean, S.; Di Guglielmo, G.M.; Philip, A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv. Wound Care 2013, 2, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Bennett, S.P.; Griffiths, G.D.; Schor, A.M.; Leese, G.P.; Schor, S.L. Growth factors in the treatment of diabetic foot ulcers. Br. J. Surg. 2003, 90, 133–146. [Google Scholar] [CrossRef]
- Badiavas, E.V.; Zhou, L.; Falanga, V. Growth inhibition of primary keratinocytes following transduction with a novel TGFbeta-1 containing retrovirus. J. Dermatol. Sci. 2001, 27, 1–6. [Google Scholar] [CrossRef]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef]
- Schmid, P.; Cox, D.; Bilbe, G.; McMaster, G.; Morrison, C.; Stahelin, H.; Luscher, N.; Seiler, W. TGF-beta s and TGF-beta type II receptor in human epidermis: Differential expression in acute and chronic skin wounds. J. Pathol. 1993, 171, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Cowin, A.J.; Hatzirodos, N.; Holding, C.A.; Dunaiski, V.; Harries, R.H.; Rayner, T.E.; Fitridge, R.; Cooter, R.D.; Schultz, G.S.; Belford, D.A. Effect of healing on the expression of transforming growth factor beta(s) and their receptors in chronic venous leg ulcers. J. Investig. Dermatol. 2001, 117, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Jude, E.B.; Blakytny, R.; Bulmer, J.; Boulton, A.J.; Ferguson, M.W. Transforming growth factor-beta 1, 2, 3 and receptor type I and II in diabetic foot ulcers. Diabet. Med. 2002, 19, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Han, G.; Owens, P.; Siddiqui, Y.; Li, A.G. Role of TGF beta-mediated inflammation in cutaneous wound healing. J. Investig. Dermatol. Symp. Proc. 2006, 11, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liarte, S.; Bernabé-García, A.; Nicolás, F.J. TGF-beta role in skin chronic wounds: a keratinocyte prespective. Cells 2020. Under review. [Google Scholar]
- Rousselle, P.; Braye, F.; Dayan, G. Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Deyrieux, A.F.; Wilson, V.G. In vitro culture conditions to study keratinocyte differentiation using the HaCaT cell line. Cytotechnology 2007, 54, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Alcaraz, A.; Mrowiec, A.; Insausti, C.L.; Bernabe-Garcia, A.; Garcia-Vizcaino, E.M.; Lopez-Martinez, M.C.; Monfort, A.; Izeta, A.; Moraleda, J.M.; Castellanos, G.; et al. Amniotic Membrane Modifies the Genetic Program Induced by TGFss, Stimulating Keratinocyte Proliferation and Migration in Chronic Wounds. PLoS ONE 2015, 10, e0135324. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta signaling through the Smad pathway: Role in extracellular matrix gene expression and regulation. J. Investig. Dermatol. 2002, 118, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, L.; Hill, C.S. Smad4 Dependency Defines Two Classes of Transforming Growth Factor (TGF-β) Target Genes and Distinguishes TGF-β -Induced Epithelial-Mesenchymal Transition from Its Antiproliferative and Migratory Responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.K.; Tomic-Canic, M.; Lucas, D.J.; Simon, M.; Blumenberg, M. TGF beta promotes the basal phenotype of epidermal keratinocytes: Transcriptional induction of K#5 and K#14 keratin genes. Growth Factors 1995, 12, 87–97. [Google Scholar] [CrossRef]
- Ramirez, H.; Patel, S.B.; Pastar, I. The Role of TGFbeta Signaling in Wound Epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Faler, B.J.; Macsata, R.A.; Plummer, D.; Mishra, L.; Sidawy, A.N. Transforming growth factor-beta and wound healing. Perspect. Vasc. Surg. Endovasc. Ther. 2006, 18, 55–62. [Google Scholar] [CrossRef]
- Bielefeld, K.A.; Amini-Nik, S.; Alman, B.A. Cutaneous wound healing: Recruiting developmental pathways for regeneration. Cell. Mol. Life Sci. 2013, 70, 2059–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedberg, I.M.; Tomic-Canic, M.; Komine, M.; Blumenberg, M. Keratins and the keratinocyte activation cycle. J. Investig. Dermatol. 2001, 116, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Landstrom, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Yang, L.; Qiu, C.X.; Ludlow, A.; Ferguson, M.W.; Brunner, G. Active transforming growth factor-beta in wound repair: Determination using a new assay. Am. J. Pathol. 1999, 154, 105–111. [Google Scholar] [CrossRef]
- Iavarone, A.; Massague, J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature 1997, 387, 417–422. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas-Terradellas, E.; Tato, I.; Bartrons, R.; Ventura, F.; Rosa, J.L. ERK and p38 pathways regulate amino acid signalling. Biochim. Biophys. Acta 2008, 1783, 2241–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abkhezr, M.; Keramati, A.R.; Ostad, S.N.; Davoodi, J.; Ghahremani, M.H. The time course of Akt and ERK activation on XIAP expression in HEK 293 cell line. Mol. Biol. Rep. 2010, 37, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gustafson-Brown, C.; Hanks, S.K.; Nason, K.; Arbeit, J.M.; Pogliano, K.; Wisdom, R.M.; Johnson, R.S. c-Jun is essential for organization of the epidermal leading edge. Dev. Cell 2003, 4, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Chen, Y.G. Regulation of TGF-beta receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Chen, Y.G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem. J. 2011, 434, 1–10. [Google Scholar] [CrossRef]
- Nicolas, F.J.; Lehmann, K.; Warne, P.H.; Hill, C.S.; Downward, J. Epithelial to Mesenchymal Transition in Madin-Darby Canine Kidney Cells Is Accompanied by Down-regulation of Smad3 Expression, Leading to Resistance to Transforming Growth Factor- beta-induced Growth Arrest. J. Biol. Chem. 2003, 278, 3251–3256. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Sarkar, P.; Mi, Q.; Wu, N.; Bringas, P.; Liu, Y.; Reddy, S.; Maxson, R.; Deng, C.; Chai, Y. Overexpression of Smad2 reveals its concerted action with Smad4 in regulating TGF-beta-mediated epidermal homeostasis. Dev. Biol. 2001, 236, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, G.S.; Yang, X.; Glick, A.B.; Weinstein, M.; Letterio, J.L.; Mizel, D.E.; Anzano, M.; Greenwell-Wild, T.; Wahl, S.M.; Deng, C.; et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999, 1, 260–266. [Google Scholar] [CrossRef]
- Ruiz-Canada, C.; Bernabé-García, A.; Liarte, S.; Insausti, C.L.; Angosto, D.; Moraleda, J.M.; Castellanos, G.; Nicolás, F.J. Amniotic membrane stimulates cell migration by modulating Transforming Growth Factor-β signaling. J. Tissue Eng. Regen. Med. 2018, 12, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Kusewitt, D.F.; Carver, E.A.; Magnino, F.; Choi, C.; Gridley, T.; Hudson, L.G. Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J. Cell. Physiol. 2005, 202, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, L.G.; Newkirk, K.M.; Chandler, H.L.; Choi, C.; Fossey, S.L.; Parent, A.E.; Kusewitt, D.F. Cutaneous wound reepithelialization is compromised in mice lacking functional Slug (Snai2). J. Dermatol. Sci. 2009, 56, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, D.S.; Blazanin, N.; Licata, M.; Lee, J.; Glick, A.B. Tumor suppressor and oncogene actions of TGFbeta1 occur early in skin carcinogenesis and are mediated by Smad3. Mol. Carcinog. 2009, 48, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, Y.; Mao, H.; Chen, W.; Luo, N.; Zhou, Q.; Chen, W.; Yu, X. A crosstalk between the Smad and JNK signaling in the TGF-beta-induced epithelial-mesenchymal transition in rat peritoneal mesothelial cells. PLoS ONE 2012, 7, e32009. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.; Haensel, D.; Gutierrez, G.; Du, H.; Dai, X.; Nie, Q. Intermediate cell states in epithelial-to-mesenchymal transition. Phys. Biol. 2019, 16, 021001. [Google Scholar] [CrossRef]
- Chen, C.R.; Kang, Y.; Siegel, P.M.; Massague, J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Warner, B.J.; Blain, S.W.; Seoane, J.; Massague, J. Myc downregulation by transforming growth factor beta required for activation of the p15(Ink4b) G(1) arrest pathway. Mol. Cell. Biol. 1999, 19, 5913–5922. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.G.; Kim, H.A.; Jong, H.S.; Park, J.H.; Kim, N.K.; Hong, S.H.; Kim, T.Y.; Bang, Y.J. The endogenous ratio of Smad2 and Smad3 influences the cytostatic function of Smad3. Mol. Biol. Cell 2005, 16, 4672–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaro, J.L.; Izzo, V.; Meaume, S.; Davies, A.H.; Lobmann, R.; Uccioli, L. Elevated levels of matrix metalloproteinases and chronic wound healing: An updated review of clinical evidence. J. Wound Care 2016, 25, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soroka, Y.; Ma’or, Z.; Leshem, Y.; Verochovsky, L.; Neuman, R.; Bregegere, F.M.; Milner, Y. Aged keratinocyte phenotyping: Morphology, biochemical markers and effects of Dead Sea minerals. Exp. Gerontol. 2008, 43, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Makrantonaki, E.; Wlaschek, M.; Scharffetter-Kochanek, K. Pathogenesis of wound healing disorders in the elderly. J. Dtsch. Dermatol. Ges. 2017, 15, 255–275. [Google Scholar] [CrossRef]
- Telgenhoff, D.; Shroot, B. Cellular senescence mechanisms in chronic wound healing. Cell Death Differ. 2005, 12, 695–698. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [Green Version]
- Vijayachandra, K.; Lee, J.; Glick, A.B. Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res. 2003, 63, 3447–3452. [Google Scholar]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Kane, C.J.; Hebda, P.A.; Mansbridge, J.N.; Hanawalt, P.C. Direct evidence for spatial and temporal regulation of transforming growth factor beta 1 expression during cutaneous wound healing. J. Cell. Physiol. 1991, 148, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Ju, W.J.; Heyer, J.; Escalante-Alcalde, D.; Stewart, C.L.; Weinstein, M.; Deng, C.; Kucherlapati, R.; Bottinger, E.P.; Roberts, A.B. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem. 2001, 276, 19945–19953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.F.; Darowish, M.; Zuscik, M.J.; Chen, D.; Schwarz, E.M.; Rosier, R.N.; Drissi, H.; O’Keefe, R.J. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 2006, 21, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, K.; Nishimoto, N.; Matsumoto, K.; Tagoh, H.; Taga, T.; Deguchi, Y.; Kuritani, T.; Hirano, T.; Hashimoto, K.; Okada, N.; et al. Interleukin 6 and expression of its receptor on epidermal keratinocytes. Cytokine 1990, 2, 381–387. [Google Scholar] [CrossRef]
- Gallucci, R.M.; Sloan, D.K.; Heck, J.M.; Murray, A.R.; O’Dell, S.J. Interleukin 6 indirectly induces keratinocyte migration. J. Investig. Dermatol. 2004, 122, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Quintero, M.; Kuri-Harcuch, W.; Gonzalez Robles, A.; Castro-Munozledo, F. Interleukin-6 promotes human epidermal keratinocyte proliferation and keratin cytoskeleton reorganization in culture. Cell Tissue Res. 2006, 325, 77–90. [Google Scholar] [CrossRef]
- Grassl, C.; Luckow, B.; Schlondorff, D.; Dendorfer, U. Transcriptional regulation of the interleukin-6 gene in mesangial cells. J. Am. Soc. Nephrol. JASN 1999, 10, 1466–1477. [Google Scholar]
- Hungness, E.S.; Pritts, T.A.; Luo, G.J.; Sun, X.; Penner, C.G.; Hasselgren, P.O. The transcription factor activator protein-1 is activated and interleukin-6 production is increased in interleukin-1beta-stimulated human enterocytes. Shock 2000, 14, 386–391. [Google Scholar] [CrossRef]
- Xie, J.; Pan, H.; Yoo, S.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 2005, 79, 15027–15037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.X.; Cao, L.P.; Kang, P.C.; Zhong, X.Y.; Lin, T.Y.; Cui, Y.F. Interleukin6 induces epithelialmesenchymal transition in human intrahepatic biliary epithelial cells. Mol. Med. Rep. 2016, 13, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Gong, Y.; Chen, Y.; Yu, D.; Wang, X.; Zhang, X.; Dou, Y.; Liu, D.; Cheng, G.; Lu, S.; et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am. J. Physiol. Ren. Physiol. 2017, 313, F310–F318. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, C.; Pan, J.; Chen, L.; Qi, S.T. Interleukin6 induces an epithelialmesenchymal transition phenotype in human adamantinomatous craniopharyngioma cells and promotes tumor cell migration. Mol. Med. Rep. 2017, 15, 4123–4131. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Seong, G.J.; Hong, S.; Jung, S.A.; Lee, J.J.; Lim, E.; Kim, S.J.; Lee, J.H. TGF-beta-induced interleukin-6 participates in transdifferentiation of human Tenon’s fibroblasts to myofibroblasts. Mol. Vis. 2009, 15, 2123–2128. [Google Scholar]
- Park, J.I.; Lee, M.G.; Cho, K.; Park, B.J.; Chae, K.S.; Byun, D.S.; Ryu, B.K.; Park, Y.K.; Chi, S.G. Transforming growth factor-beta1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene 2003, 22, 4314–4332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.L.; Topley, N.; Ito, T.; Phillips, A. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J. Biol. Chem. 2005, 280, 12239–12245. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-beta family in wound healing, burns and scarring: A review. Int. J. Burn. Trauma 2012, 2, 18–28. [Google Scholar]
- Wicks, K.; Torbica, T.; Mace, K.A. Myeloid cell dysfunction and the pathogenesis of the diabetic chronic wound. Semin. Immunol. 2014, 26, 341–353. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Human Skin Keratinocytes on Sustained TGF-β Stimulation Reveal Partial EMT Features and Weaken Growth Arrest Responses. Cells 2020, 9, 255. https://doi.org/10.3390/cells9010255
Liarte S, Bernabé-García Á, Nicolás FJ. Human Skin Keratinocytes on Sustained TGF-β Stimulation Reveal Partial EMT Features and Weaken Growth Arrest Responses. Cells. 2020; 9(1):255. https://doi.org/10.3390/cells9010255
Chicago/Turabian StyleLiarte, Sergio, Ángel Bernabé-García, and Francisco J. Nicolás. 2020. "Human Skin Keratinocytes on Sustained TGF-β Stimulation Reveal Partial EMT Features and Weaken Growth Arrest Responses" Cells 9, no. 1: 255. https://doi.org/10.3390/cells9010255