Plasma Membrane-Associated Restriction Factors and Their Counteraction by HIV-1 Accessory Proteins

 , and
, and
Abstract
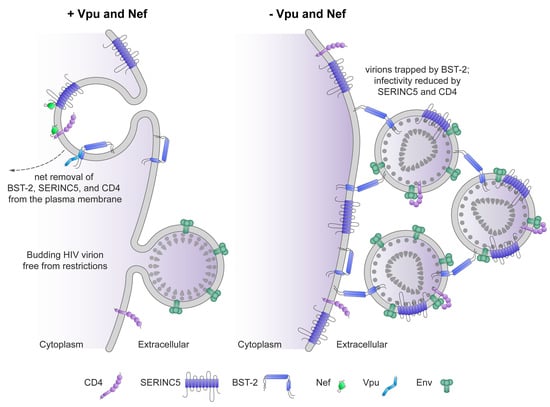
:
1. Introduction
2. The Key Plasma Membrane Proteins that Inhibit HIV-1 Release and/or Infectivity and How They Work
2.1. BST2: Historical Basis of Discovery (The Inhibitor That Vpu Counteracts to Enhance Virion-Release); Protein Topology; Mechanism of Action
2.2. SERINC Family Members: Historical Basis of Discovery (The Inhibitors That Nef Counteracts to Enhance Virion-Infectivity); Protein Topology; Mechanism of Action
2.3. CD4: The Primary Receptor, But Whose Interaction with Env During Virion-Production Inhibits Infectivity and Prematurely Exposes Epitopes on Env That Are Targets of Adaptive Immunity
3. Vpu, Nef, and the Cell Biologic Mechanisms behind Their Modulation of Membrane-Protein Trafficking
3.1. Vpu: Protein Topology, Key Interactive Surfaces and Partners (Cullin1 E3 Ubiquitin Ligase; Clathrin Adaptor Protein (AP) Complexes), Subcellular Localization, and Cellular Protein Targets
3.2. Nef: Protein Topology, Key Interactive Surfaces and Partners (AP Complexes), Subcellular Localization, and Cellular Protein Targets
3.3. The Cellular Pathways Co-Opted by Vpu and Nef
3.3.1. ERAD: Co-Opted by Vpu to Degrade CD4
3.3.2. Endocytosis: Co-Opted by Nef to Remove CD4 and SERINCs from the Plasma Membrane
3.3.3. Endo-Lysosomal Degradation
Pathways and Cofactors Co-Opted by Vpu to Degrade BST-2 (Ubiquitination, ESCRT- and AP-Complexes)
Pathways and Cofactors Co-Opted by Nef to Degrade CD4 (AP1γ2, ALIX, β-COP)
3.3.4. TGN-Retention and Block to Recycling
3.3.5. Movement within Domains of the Plasma Membrane
4. The Structural Basis of These Cell Biologic Effects: The Interactions of Vpu and Nef with Targets and Co-Factors
4.1. Nef and Vpu with CD4
4.2. Vpu with BST-2
4.3. Vpu with β-TrCP
4.4. Nef and Vpu with Clathrin AP-Complexes
5. What Else Is Known about “Global” Modulation of the Plasma Membrane by the Virus and What Is the Significance of These Cellular Targets?
5.1. Downregulated Proteins
5.2. Significance
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Daugherty, M.D.; Malik, H.S. Rules of Engagement: Molecular Insights from Host-Virus Arms Races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.D.; Plata, F. Cytotoxic T lymphocytes against HIV. AIDS 1990, 4, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.D.; Nishanian, P.; Mitsuyasu, R.; Bonavida, B. Antibody-dependent cellular cytotoxicity (ADCC)-mediated destruction of human immunodeficiency virus (HIV)-coated CD4+ T lymphocytes by acquired immunodeficiency syndrome (AIDS) effector cells. J. Clin. Immunol. 1988, 8, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef]
- Apps, R.; Del Prete, G.Q.; Chatterjee, P.; Lara, A.; Brumme, Z.L.; Brockman, M.A.; Neil, S.; Pickering, S.; Schneider, D.K.; Piechocka-Trocha, A.; et al. HIV-1 Vpu Mediates HLA-C Downregulation. Cell Host Microbe 2016, 19, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of Natural Killer Cells Following Interaction with HIV-1-Infected Cells Is Hindered by Downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, E.F.; Cohen, E.A.; Lu, Y.C.; Sodroski, J.G.; Haseltine, W.A. Functional role of human immunodeficiency virus type 1 vpu. Proc. Natl. Acad. Sci. USA 1989, 86, 5163–5167. [Google Scholar] [CrossRef] [PubMed]
- Klimkait, T.; Strebel, K.; Hoggan, M.D.; Martin, M.A.; Orenstein, J.M. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 1990, 64, 621–629. [Google Scholar] [PubMed]
- Chowers, M.Y.; Spina, C.A.; Kwoh, T.J.; Fitch, N.J.; Richman, D.D.; Guatelli, J.C. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 1994, 68, 2906–2914. [Google Scholar] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Guy, B.; Kieny, M.P.; Rivière, Y.; Le Peuch, C.; Dott, K.; Girard, M.; Montagnier, L.; Lecocq, J.-P. HIV F/3′ orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 1987, 330, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Laliena, M.; Romero, X.; March, S.; Requena, V.; Petriz, J.; Engel, P. Characterization of antibodies submitted to the B cell section of the 8th Human Leukocyte Differentiation Antigens Workshop by flow cytometry and immunohistochemistry. Cell Immunol. 2005, 236, 6–16. [Google Scholar] [CrossRef]
- Blasius, A.L.; Giurisato, E.; Cella, M.; Schreiber, R.D.; Shaw, A.S.; Colonna, M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006, 177, 3260–3265. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Viswanathan, K.; Douglas, J.L.; Hines, J.; Gustin, J.; Moses, A.V.; Früh, K. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2009, 83, 9672–9681. [Google Scholar] [CrossRef]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 2009, 83, 2382–2385. [Google Scholar] [CrossRef] [PubMed]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 Virion Entrapment by Tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.-P.; Göttlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the Bst2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Vigan, R.; Neil, S.J.D. Determinants of Tetherin Antagonism in the Transmembrane Domain of the Human Immunodeficiency Virus Type 1 Vpu Protein. J. Virol. 2010, 84, 12958–12970. [Google Scholar] [CrossRef] [Green Version]
- Skasko, M.; Wang, Y.; Tian, Y.; Tokarev, A.; Munguia, J.; Ruiz, A.; Stephens, E.B.; Opella, S.J.; Guatelli, J. HIV-1 Vpu antagonizes the innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 2012, 287, 58–67. [Google Scholar] [CrossRef]
- Rong, L.; Zhang, J.; Lu, J.; Pan, Q.; Lorgeoux, R.-P.; Aloysius, C.; Guo, F.; Liu, S.-L.; Wainberg, M.A.; Liang, C. The Transmembrane Domain of BST-2 Determines Its Sensitivity to Down-Modulation by Human Immunodeficiency Virus Type 1 Vpu. J. Virol. 2009, 83, 7536–7546. [Google Scholar] [CrossRef] [Green Version]
- Tokarev, A.A.; Munguia, J.; Guatelli, J.C. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 2011, 85, 51–63. [Google Scholar] [CrossRef]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 2014, 3, e02362. [Google Scholar] [CrossRef]
- Cocka, L.J.; Bates, P. Identification of Alternatively Translated Tetherin Isoforms with Differing Antiviral and Signaling Activities. PLoS Pathog. 2012, 8, e1002931. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.; Lukhele, S.; Hajjar, F.; Routy, J.P.; Cohen, É.A. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 2014, 11, 15. [Google Scholar] [CrossRef]
- Arias, J.F.; Heyer, L.N.; Von Bredow, B.; Weisgrau, K.L.; Moldt, B.; Burton, D.R.; Rakasz, E.G.; Evans, D.T. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 6425–6430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.X.; Barrett, B.S.; Heilman, K.J.; Messer, R.J.; Liberatore, R.A.; Bieniasz, P.D.; Kassiotis, G.; Hasenkrug, K.J.; Santiago, M.L. Tetherin promotes the innate and adaptive cell-mediated immune response against retrovirus infection in vivo. J. Immunol. 2014, 193, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Barrett, B.S.; Guo, K.; Kassiotis, G.; Hasenkrug, K.J.; Dittmer, U.; Gibbert, K.; Santiago, M.L. Tetherin/BST-2 promotes dendritic cell activation and function during acute retrovirus infection. Sci. Rep. 2016, 6, 20425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galão, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate sensing of HIV-1 assembly by tetherin induces NF-κB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.; Suarez, M.; Kwan, W.; Fitzpatrick, K.; Singh, R.; Guatelli, J. Stimulation of NF-κappaB activity by the HIV restriction factor BST2. J. Virol. 2013, 87, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Bover, L.; Cho, M.; Wen, X.; Hanabuchi, S.; Bao, M.; Rosen, D.B.; Wang, Y.-H.; Shaw, J.L.; Du, Q.; et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med. 2009, 206, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Bego, M.G.; Miguet, N.; Laliberté, A.; Aschman, N.; Gerard, F.; Merakos, A.A.; Weissenhorn, W.; Cohen, E.A. Activation of the ILT7 receptor and plasmacytoid dendritic cell responses are governed by structurally-distinct BST2 determinants. J. Boil. Chem. 2019, 294, 10503–10518. [Google Scholar] [CrossRef]
- Bego, M.G.; Côté, É.; Aschman, N.; Mercier, J.; Weissenhorn, W.; Cohen, E.A. Vpu Exploits the Cross-Talk between BST2 and the ILT7 Receptor to Suppress Anti-HIV-1 Responses by Plasmacytoid Dendritic Cells. PLoS Pathog. 2015, 11, e1005024. [Google Scholar] [CrossRef]
- Leymarie, O.; Lepont, L.; Versapuech, M.; Judith, D.; Abelanet, S.; Janvier, K.; Berlioz-Torrent, C. Contribution of the Cytoplasmic Determinants of Vpu to the Expansion of Virus-Containing Compartments in HIV-1-Infected Macrophages. J. Virol. 2019, 93, e00020-19. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an Activity-regulated Protein Family, Incorporates Serine into Membrane Lipid Synthesis. J. Boil. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed]
- Krueger, W.H.; Gonye, G.E.; Madison, D.L.; Murray, K.E.; Kumar, M.; Spoerel, N.; Pfeiffer, S.E. TPO1, a member of a novel protein family, is developmentally regulated in cultured oligodendrocytes. J. Neurochem. 1997, 69, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.R.; Luque, J.M.; Nelson, N. Identification of a ubiquitous family of membrane proteins and their expression in mouse brain. J. Exp. Boil. 2000, 203, 447–457. [Google Scholar]
- Schulte, B.; Selyutina, A.; Opp, S.; Herschhorn, A.; Sodroski, J.G.; Pizzato, M.; Diaz-Griffero, F. Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 2018, 515, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Firrito, C.; Bertelli, C.; Vanzo, T.; Chande, A.; Pizzato, M. SERINC5 as a New Restriction Factor for Human Immunodeficiency Virus and Murine Leukemia Virus. Annu. Rev. Virol. 2018, 5, 323–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Popov, S.; Göttlinger, H.G. The Nef-Like Effect of Murine Leukemia Virus Glycosylated Gag on HIV-1 Infectivity Is Mediated by Its Cytoplasmic Domain and Depends on the AP-2 Adaptor Complex. J. Virol. 2014, 88, 5900. [Google Scholar] [CrossRef]
- Pizzato, M.; Helander, A.; Popova, E.; Calistri, A.; Zamborlini, A.; Palù, G.; Göttlinger, H.G. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc. Natl. Acad. Sci. USA 2007, 104, 6812–6817. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Usami, Y.; Wu, Y.; Göttlinger, H.A. Long Cytoplasmic Loop Governs the Sensitivity of the Anti-viral Host Protein SERINC5 to HIV-1 Nef. Cell Rep. 2018, 22, 869–875. [Google Scholar] [CrossRef]
- Murrell, B.; Vollbrecht, T.; Guatelli, J.; Wertheim, J.O. The Evolutionary Histories of Antiretroviral Proteins SERINC3 and SERINC5 Do Not Support an Evolutionary Arms Race in Primates. J. Virol. 2016, 90, 8085–8089. [Google Scholar] [PubMed] [Green Version]
- Zhang, X.; Zhou, T.; Yang, J.; Lin, Y.; Shi, J.; Zhang, X.; Frabutt, D.A.; Zeng, X.; Li, S.; Venta, P.J.; et al. Identification of SERINC5-001 as the Predominant Spliced Isoform for HIV-1 Restriction. J. Virol. 2017, 91, e00137-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Lewinski, M.K.; Guatelli, J. An N-Glycosylated Form of SERINC5 Is Specifically Incorporated into HIV-1 Virions. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, A.; Bertelli, C.; Serwa, R.; Pye, V.E.; Tate, E.; Pizzato, M.; Cherepanov, P.P. Analysis of SERINC5 palmitoylation. In Retroviruses; Cold Spring Harbor: Long Island, NY, USA, 2019; p. 215. [Google Scholar]
- Stoneham, C.A.; Ramirez, P.W.; Singh, R.; Suarez, M.; Debray, A.; Lim, C.; Jia, X.; Xiong, Y.; Guatelli, J. A conserved acidic cluster motif in SERINC5 confers resistance to antagonism by HIV-1 Nef. bioRxiv 2019, 590646. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Göttlinger, H. HIV-1 Nef Responsiveness is Determined by Env Variable Regions Involved in Trimer Association and Correlates with Neutralization Sensitivity. Cell Rep. 2013, 5, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.P.; Yan, J.; Heeney, J.; McClure, M.O.; Göttlinger, H.; Luban, J.; Pizzato, M. Nef Decreases HIV-1 Sensitivity to Neutralizing Antibodies that Target the Membrane-proximal External Region of TMgp41. PLoS Pathog 2011, 7, e1002442. [Google Scholar] [CrossRef] [PubMed]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Boil. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [Green Version]
- Trautz, B.; Wiedemann, H.; Lüchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Kräusslich, H.-G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Boil. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Olety, B.; Weiss, E.R.; Popova, E.; Yamanaka, H.; Göttlinger, H. Potent Enhancement of HIV-1 Replication by Nef in the Absence of SERINC3 and SERINC5. mBio 2019, 10, e01071-19. [Google Scholar] [CrossRef] [Green Version]
- Dalgleish, A.G.; Beverley, P.C.L.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 312, 763–767. [Google Scholar] [CrossRef]
- Ross, T.M.; Oran, A.E.; Cullen, B.R. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Boil. 1999, 9, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Boil. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef]
- Stein, B.S.; Engleman, E.G. Intracellular processing of the gp160 HIV-1 envelope precursor. Endoproteolytic cleavage occurs in a cis or medial compartment of the Golgi complex. J. Boil. Chem. 1990, 265, 2640–2649. [Google Scholar]
- Willey, R.L.; Bonifacino, J.S.; Potts, B.J.; Martin, M.A.; Klausner, R.D. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. USA 1988, 85, 9580–9584. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core Structure of gp41 from the HIV Envelope Glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Mol. Boil. 1995, 2, 1075–1082. [Google Scholar] [CrossRef]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996, 85, 1135–1148. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Freed, E.O.; Myers, D.J.; Risser, R. Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc. Natl. Acad. Sci. USA 1990, 87, 4650–4654. [Google Scholar] [CrossRef]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J. Virol. 1992, 66, 226–234. [Google Scholar] [Green Version]
- Wildum, S.; Schindler, M.; Münch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 Down-Modulation and Resistance of Human Immunodeficiency Virus Type 1-Infected T Cells to Superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef] [Green Version]
- Levesque, K.; Zhao, Y.S.; Cohen, E.A. Vpu Exerts a Positive Effect on HIV-1 Infectivity by Down-modulating CD4 Receptor Molecules at the Surface of HIV-1-producing Cells. J. Boil. Chem. 2003, 278, 28346–28353. [Google Scholar] [CrossRef] [Green Version]
- Kramski, M.; Parsons, M.S.; Stratov, I.; Kent, S.J. HIV-specific antibody immunity mediated through NK cells and monocytes. Curr. HIV Res. 2013, 11, 388–406. [Google Scholar] [CrossRef]
- Forthal, D.N.; Finzi, A. Antibody-Dependent Cellular Cytotoxicity (ADCC) in HIV Infection. AIDS 2018, 32, 2439–2451. [Google Scholar] [CrossRef]
- Veillette, M.; Désormeaux, A.; Medjahed, H.; Gharsallah, N.E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, BH.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-Correlates Analysis of an HIV-1 Vaccine Efficacy Trial. New Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef]
- Bonsignori, M.; Pollara, J.; Moody, M.A.; Alpert, M.D.; Chen, X.; Hwang, K.-K.; Gilbert, P.B.; Huang, Y.; Gurley, T.C.; Kozink, D.M.; et al. Antibody-Dependent Cellular Cytotoxicity-Mediating Antibodies from an HIV-1 Vaccine Efficacy Trial Target Multiple Epitopes and Preferentially Use the VH1 Gene Family. J. Virol. 2012, 86, 11521–11532. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shi, J.; Qiu, X.; Chai, Q.; Frabutt, D.A.; Schwartz, R.C.; Zheng, Y.-H. CD4 expression and Env conformation are critical for HIV-1 restriction by SERINC5. J. Virol. 2019, 93, e00544-19. [Google Scholar] [CrossRef]
- Schwartz, S.; Felber, B.K.; Fenyö, E.M.; Pavlakis, G.N. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J. Virol. 1990, 64, 5448–5456. [Google Scholar] [Green Version]
- Zhang, H.; Lin, E.C.; Das, B.B.; Tian, Y.; Opella, S.J. Structural determination of Virus protein U from HIV-1 by NMR in membrane environments. Biochim. Biophys. Acta. 2015, 1848, 3007–3018. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Jafari, M.; Zhang, H.; Opella, S.J.; Guatelli, J. Membrane Anchoring by a C-terminal Tryptophan Enables HIV-1 Vpu to Displace Bone Marrow Stromal Antigen 2 (BST2) from Sites of Viral Assembly. J. Boil. Chem. 2015, 290, 10919–10933. [Google Scholar] [CrossRef] [Green Version]
- Schubert, U.; Ferrer-Montiel, A.V.; Oblatt-Montal, M.; Henklein, P.; Strebel, K.; Montal, M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996, 398, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Strebel, K. HIV-1 Vpu—An ion channel in search of a job. Biochim. Biophys. Acta. 2014, 1838, 1074–1081. [Google Scholar] [CrossRef]
- Schubert, U.; Henklein, P.; Boldyreff, B.; Wingender, E.; Strebel, K.; Porstmann, T. The human immunodeficiency virus type 1 encoded Vpu protein is phosphorylated by casein kinase-2 (CK-2) at positions Ser52 and Ser56 within a predicted alpha-helix-turn-alpha-helix-motif. J. Mol. Biol. 1994, 236, 16–25. [Google Scholar] [CrossRef]
- Friborg, J.; Ladha, A.; Gottlinger, H.; Haseltine, W.A.; Cohen, E.A. Functional Analysis of the Phosphorylation Sites on the Human Immunodeficiency Virus Type 1 Vpu Protein. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 8, 10. [Google Scholar] [CrossRef]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Stoneham, C.A.; Singh, R.; Jia, X.; Xiong, Y.; Guatelli, J. Endocytic Activity of HIV-1 Vpu: Phosphoserine-dependent Interactions with Clathrin Adaptors. Traffic 2017, 18, 545–561. [Google Scholar] [CrossRef]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J.D. Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS Pathog 2015, 11, e1005141. [Google Scholar] [CrossRef]
- Kueck, T.; Neil, S.J.D. A Cytoplasmic Tail Determinant in HIV-1 Vpu Mediates Targeting of Tetherin for Endosomal Degradation and Counteracts Interferon-Induced Restriction. PLoS Pathog 2012, 8, e1002609. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef]
- Magadán, J.G.; Pérez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered Mechanism of CD4 Downregulation by HIV-1 Vpu Involving Distinct ER Retention and ERAD Targeting Steps. PLoS Pathog 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Matusali, G.; Potestà, M.; Santoni, A.; Cerboni, C.; Doria, M. The Human Immunodeficiency Virus Type 1 Nef and Vpu Proteins Downregulate the Natural Killer Cell-Activating Ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, P.W.; Famiglietti, M.; Sowrirajan, B.; DePaula-Silva, A.B.; Rodesch, C.; Barker, E.; Bosque, A.; Planelles, V. Downmodulation of CCR7 by HIV-1 Vpu results in impaired migration and chemotactic signaling within CD4(+) T cells. Cell Rep. 2014, 7, 2019–2030. [Google Scholar] [CrossRef]
- Cole, G.B.; Reichheld, S.E.; Sharpe, S. FRET Analysis of the Promiscuous yet Specific Interactions of the HIV-1 Vpu Transmembrane Domain. Biophys. J. 2017, 113, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Moll, M.; Andersson, S.K.; Smed-Sörensen, A.; Sandberg, J.K. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 2010, 116, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Bächle, S.M.; Sauter, D.; Sibitz, S.; Sandberg, J.K.; Kirchhoff, F.; Moll, M. Involvement of a C-terminal motif in the interference of primate lentiviral Vpu proteins with CD1d-mediated antigen presentation. Sci. Rep. 2015, 5, 9675. [Google Scholar] [CrossRef] [Green Version]
- Bachtel, N.D.; Umviligihozo, G.; Pickering, S.; Mota, T.M.; Liang, H.; Del Prete, G.Q.; Chatterjee, P.; Lee, G.Q.; Thomas, R.; Brockman, M.A.; et al. HLA-C downregulation by HIV-1 adapts to host HLA genotype. PLoS Pathog 2018, 14, e1007257. [Google Scholar] [CrossRef]
- Yao, X.-J.; Friborg, J.; Checroune, F.; Gratton, S.; Boisvert, F.; Sékaly, R.P.; Cohen, E.A. Degradation of CD4 Induced by Human Immunodeficiency Virus Type 1 Vpu Protein: A Predicted Alpha-Helix Structure in the Proximal Cytoplasmic Region of CD4 Contributes to Vpu Sensitivity. Virology 1995, 209, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Bour, S.; Schubert, U.; Strebel, K. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: Implications for the mechanism of degradation. J. Virol. 1995, 69, 1510–1520. [Google Scholar]
- Margottin, F.; Benichou, S.; Durand, H.; Richard, V.; Liu, L.X.; Gomas, E.; Benarous, R. Interaction between the cytoplasmic domains of HIV-1 Vpu and CD4: Role of Vpu residues involved in CD4 interaction and in vitro CD4 degradation. Virology 1996, 223, 381–386. [Google Scholar] [CrossRef]
- Van Damme, N.; Guatelli, J. HIV-1 Vpu inhibits accumulation of the envelope glycoprotein within clathrin-coated, Gag-containing endosomes. Cell. Microbiol. 2008, 10, 1040–1057. [Google Scholar] [CrossRef]
- Dubé, M.; Roy, B.B.; Guiot-Guillain, P.; Mercier, J.; Binette, J.; Leung, G.; Cohen, E.A. Suppression of Tetherin-Restricting Activity upon Human Immunodeficiency Virus Type 1 Particle Release Correlates with Localization of Vpu in the trans-Golgi Network. J. Virol. 2009, 83, 4574–4590. [Google Scholar] [CrossRef]
- Jia, X.; Singh, R.; Homann, S.; Yang, H.; Guatelli, J.; Xiong, Y. Structural Basis of Evasion of Cellular Adaptive Immunity by HIV-1 Nef. Nat. Struct. Mol. Boil. 2012, 19, 701–706. [Google Scholar] [CrossRef]
- Lee, C.-H.; Saksela, K.; Mirza, U.A.; Chait, B.T.; Kuriyan, J. Crystal Structure of the Conserved Core of HIV-1 Nef Complexed with a Src Family SH3 Domain. Cell 1996, 85, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Park, S.Y.; Bonifacino, J.S.; Hurley, J.H. How HIV-1 Nef hijacks the AP-2 clathrin adaptor to downregulate CD4. eLife 2014, 3, e01754. [Google Scholar] [CrossRef]
- Craig, H.M.; Pandori, M.W.; Guatelli, J.C. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc. Natl. Acad. Sci. USA 1998, 95, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Riggs, N.L.; Craig, H.M.; Pandori, M.W.; Guatelli, J.C. The dileucine-based sorting motif in HIV-1 Nef is not required for down-regulation of class I MHC. Virology 1999, 258, 203–207. [Google Scholar] [CrossRef]
- Wonderlich, E.R.; Williams, M.; Collins, K.L. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 2008, 283, 3011–3022. [Google Scholar] [CrossRef]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Kelly, B.T.; McCoy, A.J.; Späte, K.; Miller, S.E.; Evans, P.R.; Höning, S.; Owen, D.J. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 2008, 456, 976–979. [Google Scholar] [CrossRef] [Green Version]
- Grzesiek, S.; Stahl, S.J.; Wingfield, P.T.; Bax, A. The CD4 Determinant for Downregulation by HIV-1 Nef Directly Binds to Nef. Mapping of the Nef Binding Surface by NMR. Biochemistry 1996, 35, 10256–10261. [Google Scholar] [CrossRef]
- Shu, S.T.; Emert-Sedlak, L.A.; Smithgall, T.E. Cell-based Fluorescence Complementation Reveals a Role for HIV-1 Nef Protein Dimerization in AP-2 Adaptor Recruitment and CD4 Co-receptor Down-regulation. J. Biol. Chem. 2017, 292, 2670–2678. [Google Scholar] [CrossRef] [Green Version]
- Janvier, K.; Kato, Y.; Boehm, M.; Rose, J.R.; Martina, J.A.; Kim, B.Y.; Venkatesan, S.; Bonifacino, J.S. Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 gamma-sigma1 and AP-3 delta-sigma3 hemicomplexes. J. Cell Biol. 2003, 163, 1281–1290. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Lindwasser, O.W.; Smith, W.J.; Hurley, J.H.; Bonifacino, J.S. Downregulation of CD4 by Human Immunodeficiency Virus Type 1 Nef Is Dependent on Clathrin and Involves Direct Interaction of Nef with the AP2 Clathrin Adaptor. J. Virol. 2007, 81, 3877–3890. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-H.; Thomas, L.; Ruby, C.E.; Atkins, K.M.; Morris, N.P.; Knight, Z.A.; Scholz, I.; Barklis, E.; Weinberg, A.D.; Shokat, K.M.; et al. HIV-1 Nef Assembles a Src Family Kinase-ZAP-70/Syk-PI3K Cascade to Downregulate Cell-Surface MHC-I. Cell Host Microbe 2007, 1, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Schubert, U.; Antón, L.C.; Bačík, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 Glycoprotein Degradation Induced by Human Immunodeficiency Virus Type 1 Vpu Protein Requires the Function of Proteasomes and the Ubiquitin-Conjugating Pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [Green Version]
- Magadán, J.G.; Bonifacino, J.S. Transmembrane domain determinants of CD4 Downregulation by HIV-1 Vpu. J. Virol. 2012, 86, 757–772. [Google Scholar] [CrossRef]
- Vincent, M.J.; Raja, N.U.; Jabbar, M.A. Human immunodeficiency virus type 1 Vpu protein induces degradation of chimeric envelope glycoproteins bearing the cytoplasmic and anchor domains of CD4: Role of the cytoplasmic domain in Vpu-induced degradation in the endoplasmic reticulum. J. Virol. 1993, 67, 5538–5549. [Google Scholar]
- Tiganos, E.; Yao, X.J.; Friborg, J.; Daniel, N.; Cohen, E.A. Putative alpha-helical structures in the human immunodeficiency virus type 1 Vpu protein and CD4 are involved in binding and degradation of the CD4 molecule. J. Virol. 1997, 71, 4452–4460. [Google Scholar] [Green Version]
- Jin, Y.-J.; Cai, C.Y.; Zhang, X.; Zhang, H.-T.; Hirst, J.A.; Burakoff, S.J. HIV Nef-mediated CD4 down-regulation is adaptor protein complex 2 dependent. J. Immunol. 2005, 175, 3157–3164. [Google Scholar] [CrossRef]
- Shi, J.; Xiong, R.; Zhou, T.; Su, P.; Zhang, X.; Qiu, X.; Li, H.; Li, S.; Yu, C.; Wang, B.; et al. HIV-1 Nef Antagonizes SERINC5 Restriction by Downregulation of SERINC5 via the Endosome/Lysosome System. J. Virol. 2018, 92, e00196-18. [Google Scholar] [CrossRef] [Green Version]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: Requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C.; et al. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Janvier, K.; Pelchen–Matthews, A.; Renaud, J.-B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRT-0 Component HRS is Required for HIV-1 Vpu-Mediated BST-2/Tetherin Down-Regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef]
- Ramirez, P.W.; DePaula-Silva, A.B.; Szaniawski, M.; Barker, E.; Bosque, A.; Planelles, V. HIV-1 Vpu utilizes both cullin-RING ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology 2015, 12, 9. [Google Scholar] [CrossRef]
- Tokarev, A.; Stoneham, C.; Lewinski, M.K.; Mukim, A.; Deshmukh, S.; Vollbrecht, T.; Spina, C.A.; Guatelli, J. Pharmacologic Inhibition of Nedd8 Activation Enzyme Exposes CD4-induced Epitopes within Env on Cells Expressing HIV-1. J. Virol. 2015, 90, 2486–2502. [Google Scholar] [CrossRef]
- Tavares, L.A.; da Silva, E.M.; da Silva-Januário, M.E.; Januário, Y.C.; de Cavalho, J.V.; Czernisz, E.S.; Mardones, G.A.; daSilva, L.L. CD4 downregulation by the HIV-1 protein Nef reveals distinct roles for the gamma1 and gamma2 subunits of the AP-1 complex in protein trafficking. J. Cell Sci. 2017, 130, 429–443. [Google Scholar] [CrossRef]
- Piguet, V.; Gu, F.; Foti, M.; Demaurex, N.; Gruenberg, J.; Carpentier, J.L.; Trono, D. Nef-induced CD4 degradation: A diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 1999, 97, 63–73. [Google Scholar] [CrossRef]
- Benichou, S.; Bomsel, M.; Bodéus, M.; Durand, H.; Douté, M.; Letourneur, F.; Camonis, J.; Benarous, R. Physical interaction of the HIV-1 Nef protein with beta-COP, a component of non-clathrin-coated vesicles essential for membrane traffic. J. Boil. Chem. 1994, 269, 30073–30076. [Google Scholar]
- Amorim, N.A.; Da Silva, E.M.L.; De Castro, R.O.; Da Silva-Januário, M.E.; Mendonça, L.M.; Bonifacino, J.S.; Da Costa, L.J.; DaSilva, L.L.P. Interaction of HIV-1 Nef Protein with the Host Protein Alix Promotes Lysosomal Targeting of CD4 Receptor. J. Boil. Chem. 2014, 289, 27744–27756. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.J.; Janvier, K.; Chandrasekhar, S.; Sekaly, R.P.; Bonifacino, J.S.; Venkatesan, S. CD4 down-regulation by HIV-1 and simian immunodeficiency virus (SIV) Nef proteins involves both internalization and intracellular retention mechanisms. J. Biol. Chem. 2005, 280, 7413–7426. [Google Scholar] [CrossRef]
- Schmidt, S.; Fritz, J.V.; Bitzegeio, J.; Fackler, O.T.; Keppler, O.T. HIV-1 Vpu Blocks Recycling and Biosynthetic Transport of the Intrinsic Immunity Factor CD317/Tetherin to Overcome the Virion Release Restriction. mBio 2011, 2, e00036-11. [Google Scholar] [CrossRef]
- Lau, D.; Kwan, W.; Guatelli, J. Role of the Endocytic Pathway in the Counteraction of BST-2 by Human Lentiviral Pathogens. J. Virol. 2011, 85, 9834–9846. [Google Scholar] [CrossRef] [Green Version]
- Dubé, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of Tetherin Restriction of HIV-1 Release by Vpu Involves Binding and Sequestration of the Restriction Factor in a Perinuclear Compartment. PLoS Pathog 2010, 6, e1000856. [Google Scholar] [CrossRef]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog 2013, 9. [Google Scholar] [CrossRef]
- Sharma, S.; Jafari, M.; Bangar, A.; William, K.; Guatelli, J.; Lewinski, M.K. The C-Terminal End of HIV-1 Vpu Has a Clade-Specific Determinant That Antagonizes BST-2 and Facilitates Virion Release. J. Virol. 2019, 93, e02315-18. [Google Scholar] [CrossRef]
- Rollason, R.; Dunstan, K.; Billcliff, P.G.; Bishop, P.; Gleeson, P.; Wise, H.; Digard, P.; Banting, G. Expression of HIV-1 Vpu Leads to Loss of the Viral Restriction Factor CD317/Tetherin from Lipid Rafts and Its Enhanced Lysosomal Degradation. PLoS ONE 2013, 8, e75680. [Google Scholar] [CrossRef]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Müller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 Vpu Antagonizes CD317/Tetherin by Adaptor Protein-1-Mediated Exclusion from Virus Assembly Sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef] [Green Version]
- Preusser, A.; Briese, L.; Willbold, D. Presence of a Helix in Human CD4 Cytoplasmic Domain Promotes Binding to HIV-1 Nef Protein. Biochem. Biophys. Res. Commun. 2002, 292, 734–740. [Google Scholar] [CrossRef]
- Garcia, J.V.; Miller, A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef]
- Manrique, S.; Sauter, D.; Horenkamp, F.A.; Lülf, S.; Yu, H.; Hotter, D.; Anand, K.; Kirchhoff, F.; Geyer, M. Endocytic sorting motif interactions involved in Nef-mediated downmodulation of CD4 and CD3. Nat. Commun. 2017, 8, 442. [Google Scholar] [CrossRef] [Green Version]
- McNatt, M.W.; Zang, T.; Hatziioannou, T.; Bartlett, M.; Ben Fofana, I.; Johnson, W.E.; Neil, S.J.D.; Bieniasz, P.D. Species-Specific Activity of HIV-1 Vpu and Positive Selection of Tetherin Transmembrane Domain Variants. PLoS Pathog 2009, 5, e1000300. [Google Scholar] [CrossRef]
- Wu, G.; Xu, G.; Schulman, B.A.; Jeffrey, P.D.; Harper, J.W.; Pavletich, N.P. Structure of a beta-TrCP1-Skp1-beta-catenin complex: Destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol. Cell 2003, 11, 1445–1456. [Google Scholar] [CrossRef]
- Evrard-Todeschi, N.; Gharbi-Benarous, J.; Bertho, G.; Coadou, G.; Megy, S.; Benarous, R.; Girault, J.P. NMR studies for identifying phosphopeptide ligands of the HIV-1 protein Vpu binding to the F-box protein beta-TrCP. Peptides 2006, 27, 194–210. [Google Scholar] [CrossRef]
- Ahle, S.; Mann, A.; Eichelsbacher, U.; Ungewickell, E. Structural relationships between clathrin assembly proteins from the Golgi and the plasma membrane. EMBO J. 1988, 7, 919–929. [Google Scholar] [CrossRef]
- Robinson, M.S. Immunofluorescent localization of 100K coated vesicle proteins. J. Cell Boil. 1986, 102, 48–54. [Google Scholar] [CrossRef]
- Hirst, J.; Robinson, M.S. Clathrin and adaptors. Biochim. Biophys. Acta. 1998, 1404, 173–193. [Google Scholar] [CrossRef] [Green Version]
- Keen, J.H. Clathrin and Associated Assembly and Disassembly Proteins. Annu. Rev. Biochem. 1990, 59, 415–438. [Google Scholar] [CrossRef]
- Pearse, B.M.; Robinson, M.S. Clathrin, adaptors, and sorting. Annu. Rev. Cell Biol. 1990, 6, 151–171. [Google Scholar] [CrossRef]
- Schmid, S.L. Clathrin-Coated Vesicle Formation and Protein Sorting: An Integrated Process. Annu. Rev. Biochem. 1997, 66, 511–548. [Google Scholar] [CrossRef]
- Singh, R.; Stoneham, C.; Lim, C.; Jia, X.; Guenaga, J.; Wyatt, R.; Wertheim, J.O.; Xiong, Y.; Guatelli, J. Phosphoserine acidic cluster motifs bind distinct basic regions on the mu subunits of clathrin adaptor protein complexes. J. Biol. Chem. 2018, 293, 15678–15690. [Google Scholar] [CrossRef]
- Navarro Negredo, P.; Edgar, J.R.; Wrobel, A.G.; Zaccai, N.R.; Antrobus, R.; Owen, D.J.; Robinson, M.S.; Navarro Negredo, P.; et al. Contribution of the clathrin adaptor AP-1 subunit micro1 to acidic cluster protein sorting. J. Cell Biol. 2017, 216, 2927–2943. [Google Scholar]
- Haller, C.; Müller, B.; Fritz, J.V.; Lamas-Murua, M.; Stolp, B.; Pujol, F.M.; Keppler, O.T.; Fackler, O.T. HIV-1 Nef and Vpu Are Functionally Redundant Broad-Spectrum Modulators of Cell Surface Receptors, Including Tetraspanins. J. Virol. 2014, 88, 14241–14257. [Google Scholar] [CrossRef] [Green Version]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Boso, G.; Langer, S.; Soonthornvacharin, S.; De Jesus, P.D.; Nguyen, Q.; Olivieri, K.C.; Portillo, A.J.; Yoh, S.M.; Pache, L.; et al. Large-Scale Arrayed Analysis of Protein Degradation Reveals Cellular Targets for HIV-1 Vpu. Cell Rep. 2018, 22, 2493–2503. [Google Scholar] [CrossRef] [Green Version]
- Sugden, S.M.; Pham, T.N.Q.; Cohen, E.A. HIV-1 Vpu Downmodulates ICAM-1 Expression, Resulting in Decreased Killing of Infected CD4(+) T Cells by NK Cells. J. Virol. 2017, 91, e02442-16. [Google Scholar] [CrossRef]
- Enard, D.; Cai, L.; Gwennap, C.; Petrov, D.A. Viruses are a dominant driver of protein adaptation in mammals. eLife 2016, 5, 56. [Google Scholar] [CrossRef]
- Laguette, N.; Rahm, N.; Sobhian, B.; Chable-Bessia, C.; Münch, J.; Snoeck, J.; Sauter, D.; Switzer, W.M.; Heneine, W.; Kirchhoff, F.; et al. Evolutionary and Functional Analyses of the Interaction between the Myeloid Restriction Factor SAMHD1 and the Lentiviral Vpx Protein. Cell Host Microbe 2012, 11, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.S.; Malik, H.S.; Emerman, M. Ancient Adaptive Evolution of Tetherin Shaped the Functions of Vpu and Nef in Human Immunodeficiency Virus and Primate Lentiviruses. J. Virol. 2010, 84, 7124–7134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient Adaptive Evolution of the Primate Antiviral DNA-Editing Enzyme APOBEC3G. PLoS Boil. 2004, 2, e275. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Serra-Moreno, R.; Neidermyer, W.; Rahmberg, A.; Mackey, J.; Ben Fofana, I.; Johnson, W.E.; Westmoreland, S.; Evans, D.T. Species-Specific Activity of SIV Nef and HIV-1 Vpu in Overcoming Restriction by Tetherin/BST2. PLoS Pathog 2009, 5, e1000429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wilson, S.J.; Langford, W.; Virgen, B.; Gregory, D.; Johnson, M.C.; Münch, J.; Kirchhoff, F.; Bieniasz, P.D.; Hatziioannou, T. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 2009, 6, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Nydegger, S. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Exp. Med. 2006, 203, 795–807. [Google Scholar] [CrossRef]
- Lambele, M.; Koppensteiner, H.; Symeonides, M.; Roy, N.H.; Chan, J.; Schindler, M.; Thali, M. Vpu Is the Main Determinant for Tetraspanin Downregulation in HIV-1-Infected Cells. J. Virol. 2015, 89, 3247–3255. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Waheed, A.A.; Yu, J.; Zeng, C.; Chen, H.Y.; Zheng, Y.M.; Feizpour, A.; Reinhard, B.M.; Gummuluru, S.; Lin, S.; et al. TIM-mediated inhibition of HIV-1 release is antagonized by Nef but potentiated by SERINC proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 5705–5714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerson, N.R.; Rowley, P.A.; Swan, C.H.; Le, D.T.; Wilkerson, G.K.; Sawyer, S.L.; Wilkerson, G.K. Positive selection of primate genes that promote HIV-1 replication. Virology 2014, 454, 291–298. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Downregulation/Degradation | Binding | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Modulated by | Cellular Adaptor Utilized by Vpu or Nef | Counteraction Mechanism | Domains or Residues Required | Domains or Residues Required | ||||||||||
| Host PM Protein | Biological Function | Nef | Vpu | AP-1 | AP-2 | SCFb-TrCP | Proteasomal Degradation | Lysosomal Degradation | Endosomal Sequestration | Nef | Vpu | Nef | Vpu | Reference(s) (PMID#) |
| BST-2 (CD317) | ISG: Traps enveloped viral particles on plasma membrane | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | N/A | TM (A10,A14,18,W22), S52,56; 59ExxxLV | N/A | TM (A10,A14,18,W22) | 18200009; 18342597 | ||
| CCR5 (CD195) | Chemokine receptor: inflammatory response | ✓ | ✓ | ✓ | G2; 62EEEE; PxxP | Unknown | N/A | 15854903; 26178998 | ||||||
| CCR7 (CD197) | Homing receptor: recruitment of immune cells to lymphoid tissues | ✓ | ✓ | N/A | TM (A10, A14, A18, W22) | N/A | Unknown | 24910430 | ||||||
| CD1d | APC: Present lipid antigens to NKT cells | ✓ | ✓ | ✓ | Unknown | CD (APW76) cladeB | 15916790; 16385629; 20530791; 25872908 | |||||||
| CD28 | TCR complex: co-stimulation, activation | ✓ | ✓ | ✓ | LL165; DD175 | 59ExxxLV; S52,56 | DD175 | S52,56 | 29329537 | |||||
| CD4 | TCR co-receptor: T cell activation; HIV-1 Env receptor | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | G2; 57WL; G95; G96; L97; R106; L110; 160ExxxLL; 174DD | TM; L63; V68; S52, 56 | 57WL, G95, G96,L97, R106, L110 | TM; CD | 3118220; 1433512 | ||
| CD62L | Leukocyte adhesion and signaling | ✓ | ✓ | ✓ | Unknown | Unknown | Unknown | Unknown | 25822027 | |||||
| CXCR4 (CD184) | Chemokine receptor: inflammatory response | ✓ | ✓ | 62EEEE; PxxP | Unknown | N/A | 16928758 | |||||||
| ICAM-1 (CD54) | Leukocyte adhesion; NK cell activation | ✓ | ✓ | ✓ | N/A | TM (A10,A14, A18); S52,56 | TM (A10,A14, A18) | 28148794 | ||||||
| MHC-I | Induction of Adaptive Immunity (CD8+ T cells): Antigen presentation to APCs | ✓ HLA-A/B | ✓ HLA-C | ✓ | ✓ | W13; R17; R19; M20; 62EEEE; P78; W113; Y120; D123 | TM (LE5; L16; L18 in WITO) | Tri-molecular complex with AP-1 (W13, M20, 62EEEE, P78, D123) | Unknown | 8612235; 9450757; 22705789; 27173934 | ||||
| MHC-II | Induction of Adaptive Immunity (CD4+ T cells): Antigen presentation to APCs | ✓ | 62EEEE; P75; P78; LL164,165 | N/A | N/A | 11593029 | ||||||||
| NKG2D-L | Activation receptor: induction of NK cell mediated cytotoxicity and cytokine release | ✓ | G2 | N/A | N/A | 17170457; 19424050 | ||||||||
| NTB-A (CD352) | Co-activation receptor: induction of NK cell mediated cytotoxicity and cytokine release | ✓ | ✓ | N/A | TM (A18) | N/A | TM | 21075351 | ||||||
| PVR (CD155) | Activation receptor: induction of NK cell mediated cytotoxicity and cytokine release | ✓ | ✓ | ✓ | ✓ | 72PxxPxxP; 62EEEE; F191 | TM (A10,A14, A18), S52,56 | Unknown | TM (A10,A14, A18) | 22301152; 25113908 | ||||
| SERINC3/5 | Phospholipid biosynthesis; Reduce retroviral infectivity | ✓ | ✓ | ✓ | ✓ | G2; CAW57; D123; LL165; ED175 | I109; L112; W115; F121 | 26416734; 29514909; 27681140 | ||||||
| SNAT1 | Immunometabolism (amino acid - alanine - transporter) | ✓ | ✓ | ✓ | S52,56; TM (W22) | Unknown | 26439863 | |||||||
| Tetraspanins | Membrane organization | ✓ | ✓ | ✓ | Variable | S52,56; TM (partial) | Unknown | Unknown | 25275127; 25568205 | |||||
| CD99 | PM T cell receptor: Regulator of focal adhesions, cell-cell junctions | ✓ | N/A | Unknown | Unknown | Unknown | 29490283 | |||||||
| PLP2 | Membrane trafficking | ✓ | N/A | Unknown | Unknown | Unknown | 29490283 | |||||||
| TIM-1 (CD365) | T-cell activation, cellular proliferation, apoptosis, immune tolerance | ✓ | ✓ | ✓ | G2; D123, LL165 | N/A | Unknown | Unknown | 30842281 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, P.W.; Sharma, S.; Singh, R.; Stoneham, C.A.; Vollbrecht, T.; Guatelli, J. Plasma Membrane-Associated Restriction Factors and Their Counteraction by HIV-1 Accessory Proteins. Cells 2019, 8, 1020. https://doi.org/10.3390/cells8091020
Ramirez PW, Sharma S, Singh R, Stoneham CA, Vollbrecht T, Guatelli J. Plasma Membrane-Associated Restriction Factors and Their Counteraction by HIV-1 Accessory Proteins. Cells. 2019; 8(9):1020. https://doi.org/10.3390/cells8091020
Chicago/Turabian StyleRamirez, Peter W., Shilpi Sharma, Rajendra Singh, Charlotte A. Stoneham, Thomas Vollbrecht, and John Guatelli. 2019. "Plasma Membrane-Associated Restriction Factors and Their Counteraction by HIV-1 Accessory Proteins" Cells 8, no. 9: 1020. https://doi.org/10.3390/cells8091020