Targeting the CD40-CD154 Signaling Pathway for Treatment of Autoimmune Arthritis


Abstract
:
1. Introduction
2. CD154 and the CD40-CD154 Interaction
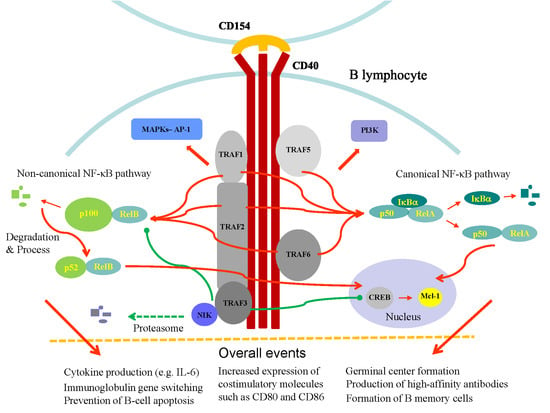
3. CD40 and CD40-Mediated Signaling
4. The CD40-CD154 Interaction in the Pathogenesis of Autoimmune Disorders
5. Clinical Analyses of Blocking the CD40-CD154 Axis in Humans
5.1. Anti-CD154
5.2. Anti-CD40
6. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- O’Dell, J.R.; Mikuls, T.R.; Taylor, T.H.; Ahluwalia, V.; Brophy, M.; Warren, S.R.; Lew, R.A.; Cannella, A.C.; Kunkel, G.; Phibbs, C.S.; et al. Therapies for active rheumatoid arthritis after methotrexate failure. N. Engl. J. Med. 2013, 369, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. T-cell-targeted therapies in rheumatoid arthritis. Nat. Clin. Pr. Rheumatol. 2006, 2, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells 2018, 7, 223. [Google Scholar] [CrossRef] [PubMed]
- Pilat, N.; Sayegh, M.H.; Wekerle, T. Costimulatory pathways in transplantation. Semin. Immunol. 2011, 23, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai-Mehta, A.; Lu, L.; Ramsey-Goldman, R.; Datta, S.K. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J. Clin. Invest. 1996, 97, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Berger, D.; Crow, M.K. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J. Clin. Invest. 1996, 98, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Daoussis, D.; Antonopoulos, I.; Andonopoulos, A.P.; Liossis, S.N. Increased expression of CD154 (CD40L) on stimulated T-cells from patients with psoriatic arthritis. Rheumatology 2007, 46, 227–231. [Google Scholar] [CrossRef]
- Berner, B.; Wolf, G.; Hummel, K.M.; Muller, G.A.; Reuss-Borst, M.A. Increased expression of CD40 ligand (CD154) on CD4+ T cells as a marker of disease activity in rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Tung, C.H.; Lu, M.C.; Lai, N.S.; Wu, S.F. Tumor necrosis factor-alpha blockade treatment decreased CD154 (CD40-ligand) expression in rheumatoid arthritis. PLoS ONE 2017, 12, e0183726. [Google Scholar] [CrossRef]
- Ranheim, E.A.; Kipps, T.J. Elevated expression of CD80 (B7/BB1) and other accessory molecules on synovial fluid mononuclear cell subsets in rheumatoid arthritis. Arthritis Rheum. 1994, 37, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, H.; Kawaguchi, Y.; Harigai, M.; Hara, M.; Saito, S.; Yamaguchi, T.; Shimada, K.; Kawamoto, M.; Tomatsu, T.; Kamatani, N. Increased CD40 expression on articular chondrocytes from patients with rheumatoid arthritis: Contribution to production of cytokines and matrix metalloproteinases. J. Rheumatol. 2004, 31, 1506–1512. [Google Scholar] [PubMed]
- Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J.F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front Immunol. 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Wang, M.; Chen, L.; Guo, Y.; Chen, Z.; Wu, J. Increased expression of CD40/TRAF1 and activation of nuclear factor-kappakappaB-dependent proinflammatory gene expression in collagen-induced arthritis. Scand. J. Rheumatol. 2018, 47, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Rissoan, M.C.; Van Kooten, C.; Chomarat, P.; Galibert, L.; Durand, I.; Thivolet-Bejui, F.; Miossec, P.; Banchereau, J. The functional CD40 antigen of fibroblasts may contribute to the proliferation of rheumatoid synovium. Clin. Exp. Immunol. 1996, 106, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sakurai, J.; Ohata, J.; Kohsaka, T.; Hashimoto, H.; Okumura, K.; Abe, R.; Azuma, M. Involvement of CD40 ligand-CD40 and CTLA4-B7 pathways in murine acute graft-versus-host disease induced by allogeneic T cells lacking CD28. J. Immunol. 1998, 160, 4225–4231. [Google Scholar] [PubMed]
- Jansen, M.F.; Hollander, M.R.; van Royen, N.; Horrevoets, A.J.; Lutgens, E. CD40 in coronary artery disease: A matter of macrophages? Basic Res. Cardiol. 2016, 111, 38. [Google Scholar] [CrossRef]
- Fisher, M.Z.; Fisher, B.; Ng, W.-F.; Bombardieri, M.; Posch, M.; Papas, A.S.; Farag, A.M.; Daikeler, T.; Bannert, B.; Kivitz, A.J.; et al. The Novel Anti-CD40 Monoclonal Antibody CFZ533 Shows Beneficial Effects in Patients with Primary Sjögren’s Syndrome: A Phase IIa Double-Blind, Placebo-Controlled Randomized Trial, American College of Rheumatology. 2017. Available online: https://cdn.website.thryv.com/a9ef1d53e14a4fbd9aab83a0e76b3fc1/files/uploaded/acrabstracts2017%231784.pdf (accessed on 18 September 2017).
- Boleto, G.; Allanore, Y.; Avouac, J. Targeting Costimulatory Pathways in Systemic Sclerosis. Front Immunol. 2018, 9, 2998. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, M.; Nomura, S.; Fujimura, K.; Nagasawa, T.; Muto, Y.; Kurata, Y.; Tanaka, S.; Ikeda, Y. Effect of a single injection of humanized anti-CD154 monoclonal antibody on the platelet-specific autoimmune response in patients with immune thrombocytopenic purpura. Blood 2004, 103, 1229–1236. [Google Scholar] [CrossRef]
- Senhaji, N.; Kojok, K.; Darif, Y.; Fadainia, C.; Zaid, Y. The Contribution of CD40/CD40L Axis in Inflammatory Bowel Disease: An Update. Front Immunol. 2015, 6, 529. [Google Scholar] [CrossRef]
- Croft, M.; Siegel, R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 217–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, R.J.; Fanslow, W.C.; Strockbine, L.; Sato, T.A.; Clifford, K.N.; Macduff, B.M.; Anderson, D.M.; Gimpel, S.D.; Davis-Smith, T.; Maliszewski, C.R.; et al. Molecular and biological characterization of a murine ligand for CD40. Nature 1992, 357, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.A.; Zirlik, A.; Wolf, D. CD40L and Its Receptors in Atherothrombosis-An Update. Front Cardiovasc. Med. 2017, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Biram, A.; Davidzohn, N.; Shulman, Z. T cell interactions with B cells during germinal center formation, a three-step model. Immunol. Rev. 2019, 288, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Flavell, R.A. A central role of CD40 ligand in the regulation of CD4+ T-cell responses. Immunol. Today 1996, 17, 410–414. [Google Scholar] [CrossRef]
- Yacoub, D.; Benslimane, N.; Al-Zoobi, L.; Hassan, G.; Nadiri, A.; Mourad, W. CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J. Biol. Chem. 2013, 288, 36083–36093. [Google Scholar] [CrossRef] [PubMed]
- Pietravalle, F.; Lecoanet-Henchoz, S.; Blasey, H.; Aubry, J.P.; Elson, G.; Edgerton, M.D.; Bonnefoy, J.Y.; Gauchat, J.F. Human native soluble CD40L is a biologically active trimer, processed inside microsomes. J. Biol. Chem. 1996, 271, 5965–5967. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.M.; Lucci, J.; Su, L.; Ehrenfels, B.; Garber, E.; Thomas, D. Heteromultimeric complexes of CD40 ligand are present on the cell surface of human T lymphocytes. J. Biol. Chem. 1997, 272, 911–915. [Google Scholar] [CrossRef]
- Miller, E.A.; Gopal, R.; Valdes, V.; Berger, J.S.; Bhardwaj, N.; O’Brien, M.P. Soluble CD40 ligand contributes to dendritic cell-mediated T-cell dysfunction in HIV-1 infection. AIDS 2015, 29, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Fanslow, W.C.; Srinivasan, S.; Paxton, R.; Gibson, M.G.; Spriggs, M.K.; Armitage, R.J. Structural characteristics of CD40 ligand that determine biological function. Semin. Immunol. 1994, 6, 267–278. [Google Scholar] [CrossRef]
- Haswell, L.E.; Glennie, M.J.; Al-Shamkhani, A. Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur. J. Immunol. 2001, 31, 3094–3100. [Google Scholar] [CrossRef]
- Rao, D.A. T Cells That Help B Cells in Chronically Inflamed Tissues. Front Immunol. 2018, 9, 1924. [Google Scholar] [CrossRef]
- Monaco, C.; Andreakos, E.; Young, S.; Feldmann, M.; Paleolog, E. T cell-mediated signaling to vascular endothelium: Induction of cytokines, chemokines, and tissue factor. J. Leukoc. Biol. 2002, 71, 659–668. [Google Scholar]
- Cho, C.S.; Cho, M.L.; Min, S.Y.; Kim, W.U.; Min, D.J.; Lee, S.S.; Park, S.H.; Choe, J.; Kim, H.Y. CD40 engagement on synovial fibroblast up-regulates production of vascular endothelial growth factor. J. Immunol. 2000, 164, 5055–5061. [Google Scholar] [CrossRef]
- Guo, Y.; Walsh, A.M.; Fearon, U.; Smith, M.D.; Wechalekar, M.D.; Yin, X.; Cole, S.; Orr, C.; McGarry, T.; Canavan, M.; et al. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J. Immunol. 2017, 198, 4490–4501. [Google Scholar] [CrossRef] [Green Version]
- Mewar, D.; Wilson, A.G. Autoantibodies in rheumatoid arthritis: A review. Biomed. Pharm. 2006, 60, 648–655. [Google Scholar] [CrossRef]
- Sharpe, A.H. Mechanisms of costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Stout, R.D.; Suttles, J. The many roles of CD40 in cell-mediated inflammatory responses. Immunol. Today 1996, 17, 487–492. [Google Scholar] [CrossRef]
- Hollenbaugh, D.; Mischel-Petty, N.; Edwards, C.P.; Simon, J.C.; Denfeld, R.W.; Kiener, P.A.; Aruffo, A. Expression of functional CD40 by vascular endothelial cells. J. Exp. Med. 1995, 182, 33–40. [Google Scholar] [CrossRef]
- Fries, K.M.; Sempowski, G.D.; Gaspari, A.A.; Blieden, T.; Looney, R.J.; Phipps, R.P. CD40 expression by human fibroblasts. Clin. Immunol. Immunopathol. 1995, 77, 42–51. [Google Scholar] [CrossRef]
- Karmann, K.; Hughes, C.C.; Schechner, J.; Fanslow, W.C.; Pober, J.S. CD40 on human endothelial cells: Inducibility by cytokines and functional regulation of adhesion molecule expression. Proc. Natl. Acad. Sci. USA 1995, 92, 4342–4346. [Google Scholar] [CrossRef]
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Schonbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell Mol. Life Sci. 2001, 58, 4–43. [Google Scholar]
- Foy, T.M.; Laman, J.D.; Ledbetter, J.A.; Aruffo, A.; Claassen, E.; Noelle, R.J. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J. Exp. Med. 1994, 180, 157–163. [Google Scholar] [CrossRef]
- Kawabe, T.; Naka, T.; Yoshida, K.; Tanaka, T.; Fujiwara, H.; Suematsu, S.; Yoshida, N.; Kishimoto, T.; Kikutani, H. The immune responses in CD40-deficient mice: Impaired immunoglobulin class switching and germinal center formation. Immunity 1994, 1, 167–178. [Google Scholar] [CrossRef]
- Bishop, G.A.; Hostager, B.S. Signaling by CD40 and its mimics in B cell activation. Immunol. Res. 2001, 24, 97–109. [Google Scholar] [CrossRef]
- Rothe, M.; Wong, S.C.; Henzel, W.J.; Goeddel, D.V. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell 1994, 78, 681–692. [Google Scholar] [CrossRef]
- Park, H.H. Structure of TRAF Family: Current Understanding of Receptor Recognition. Front Immunol. 2018, 9, 1999. [Google Scholar] [CrossRef] [Green Version]
- Bishop, G.A.; Hostager, B.S. The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. 2003, 14, 297–309. [Google Scholar] [CrossRef]
- Kraus, Z.J.; Nakano, H.; Bishop, G.A. TRAF5 is a critical mediator of in vitro signals and in vivo functions of LMP1, the viral oncogenic mimic of CD40. Proc. Natl. Acad. Sci. USA 2009, 106, 17140–17145. [Google Scholar] [CrossRef] [Green Version]
- Bishop, G.A.; Moore, C.R.; Xie, P.; Stunz, L.L.; Kraus, Z.J. TRAF proteins in CD40 signaling. Adv. Exp. Med. Biol. 2007, 597, 131–151. [Google Scholar]
- Dejardin, E. The alternative NF-kappaB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharm. 2006, 72, 1161–1179. [Google Scholar] [CrossRef]
- Shih, V.F.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef]
- Lee, H.Y.; Jeon, H.S.; Song, E.K.; Han, M.K.; Park, S.I.; Lee, S.I.; Yun, H.J.; Kim, J.R.; Kim, J.S.; Lee, Y.C.; et al. CD40 ligation of rheumatoid synovial fibroblasts regulates RANKL-mediated osteoclastogenesis: Evidence of NF-kappaB-dependent, CD40-mediated bone destruction in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 1747–1758. [Google Scholar] [CrossRef]
- Schonbeck, U.; Libby, P. CD40 signaling and plaque instability. Circ. Res. 2001, 89, 1092–1103. [Google Scholar] [CrossRef]
- Wolf, D.; Jehle, F.; Michel, N.A.; Bukosza, E.N.; Rivera, J.; Chen, Y.C.; Hoppe, N.; Dufner, B.; Rodriguez, A.O.; Colberg, C.; et al. Coinhibitory suppression of T cell activation by CD40 protects against obesity and adipose tissue inflammation in mice. Circulation 2014, 129, 2414–2425. [Google Scholar] [CrossRef]
- Pullen, S.S.; Dang, T.T.; Crute, J.J.; Kehry, M.R. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J. Biol. Chem. 1999, 274, 14246–14254. [Google Scholar] [CrossRef]
- Xie, P.; Hostager, B.S.; Munroe, M.E.; Moore, C.R.; Bishop, G.A. Cooperation between TNF receptor-associated factors 1 and 2 in CD40 signaling. J. Immunol. 2006, 176, 5388–5400. [Google Scholar] [CrossRef]
- Arron, J.R.; Pewzner-Jung, Y.; Walsh, M.C.; Kobayashi, T.; Choi, Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J. Exp. Med. 2002, 196, 923–934. [Google Scholar] [CrossRef]
- Carpentier, I.; Beyaert, R. TRAF1 is a TNF inducible regulator of NF-kappaB activation. Febs. Lett. 1999, 460, 246–250. [Google Scholar] [CrossRef]
- Leo, E.; Deveraux, Q.L.; Buchholtz, C.; Welsh, K.; Matsuzawa, S.; Stennicke, H.R.; Salvesen, G.S.; Reed, J.C. TRAF1 is a substrate of caspases activated during tumor necrosis factor receptor-alpha-induced apoptosis. J. Biol. Chem. 2001, 276, 8087–8093. [Google Scholar] [CrossRef]
- Duckett, C.S.; Gedrich, R.W.; Gilfillan, M.C.; Thompson, C.B. Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol. Cell Biol. 1997, 17, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Edilova, M.I.; Abdul-Sater, A.A.; Watts, T.H. TRAF1 Signaling in Human Health and Disease. Front Immunol. 2018, 9, 2969. [Google Scholar] [CrossRef] [Green Version]
- Rowland, S.L.; Tremblay, M.M.; Ellison, J.M.; Stunz, L.L.; Bishop, G.A.; Hostager, B.S. A novel mechanism for TNFR-associated factor 6-dependent CD40 signaling. J. Immunol. 2007, 179, 4645–4653. [Google Scholar] [CrossRef]
- Yao, Y.; Huang, W.; Li, X.; Li, X.; Qian, J.; Han, H.; Sun, H.; An, X.; Lu, L.; Zhao, H. Tespa1 Deficiency Dampens Thymus-Dependent B-Cell Activation and Attenuates Collagen-Induced Arthritis in Mice. Front Immunol. 2018, 9, 965. [Google Scholar] [CrossRef]
- Seibold, K.; Ehrenschwender, M. p62 regulates CD40-mediated NFkappaB activation in macrophages through interaction with TRAF6. Biochem. Biophys. Res. Commun. 2015, 464, 330–335. [Google Scholar] [CrossRef]
- Seijkens, T.T.P.; van Tiel, C.M.; Kusters, P.J.H.; Atzler, D.; Soehnlein, O.; Zarzycka, B.; Aarts, S.; Lameijer, M.; Gijbels, M.J.; Beckers, L.; et al. Targeting CD40-Induced TRAF6 Signaling in Macrophages Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 527–542. [Google Scholar] [CrossRef]
- Bushell, K.R.; Kim, Y.; Chan, F.C.; Ben-Neriah, S.; Jenks, A.; Alcaide, M.; Fornika, D.; Grande, B.M.; Arthur, S.; Gascoyne, R.D.; et al. Genetic inactivation of TRAF3 in canine and human B-cell lymphoma. Blood 2015, 125, 999–1005. [Google Scholar] [CrossRef]
- Malinin, N.L.; Boldin, M.P.; Kovalenko, A.V.; Wallach, D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature 1997, 385, 540–544. [Google Scholar] [CrossRef]
- Yin, L.; Wu, L.; Wesche, H.; Arthur, C.D.; White, J.M.; Goeddel, D.V.; Schreiber, R.D. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science 2001, 291, 2162–2165. [Google Scholar] [CrossRef]
- Thu, Y.M.; Richmond, A. NF-kappaB inducing kinase: A key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010, 21, 213–226. [Google Scholar] [CrossRef]
- Liao, G.; Zhang, M.; Harhaj, E.W.; Sun, S.C. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004, 279, 26243–26250. [Google Scholar] [CrossRef]
- Xie, P.; Stunz, L.L.; Larison, K.D.; Yang, B.; Bishop, G.A. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 2007, 27, 253–267. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Mambetsariev, N.; Lin, W.W.; Stunz, L.L.; Hanson, B.M.; Hildebrand, J.M.; Bishop, G.A. Nuclear TRAF3 is a negative regulator of CREB in B cells. Proc. Natl. Acad. Sci. USA 2016, 113, 1032–1037. [Google Scholar] [CrossRef] [Green Version]
- Bishop, G.A.; Stunz, L.L.; Hostager, B.S. TRAF3 as a Multifaceted Regulator of B Lymphocyte Survival and Activation. Front Immunol. 2018, 9, 2161. [Google Scholar] [CrossRef]
- Early, G.S.; Zhao, W.; Burns, C.M. Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand black x New Zealand white mice. Response correlates with the absence of an anti-antibody response. J. Immunol. 1996, 157, 3159–3164. [Google Scholar]
- Mohan, C.; Shi, Y.; Laman, J.D.; Datta, S.K. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J. Immunol. 1995, 154, 1470–1480. [Google Scholar]
- Perper, S.J.; Westmoreland, S.V.; Karman, J.; Twomey, R.; Seagal, J.; Wang, R.; McRae, B.L.; Clarke, S.H. Treatment with a CD40 Antagonist Antibody Reverses Severe Proteinuria and Loss of Saliva Production and Restores Glomerular Morphology in Murine Systemic Lupus Erythematosus. J. Immunol. 2019, 203, 58–75. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Suzuki, M.; Zhang, X.; Ichim, T.E.; Zhu, F.; Ling, H.; Shunnar, A.; Wang, M.H.; Garcia, B.; Inman, R.D.; et al. RNAi-mediated CD40-CD154 interruption promotes tolerance in autoimmune arthritis. Arthritis Res. 2010, 12, R13. [Google Scholar] [CrossRef]
- Durie, F.H.; Fava, R.A.; Foy, T.M.; Aruffo, A.; Ledbetter, J.A.; Noelle, R.J. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science 1993, 261, 1328–1330. [Google Scholar] [CrossRef]
- Tellander, A.C.; Michaelsson, E.; Brunmark, C.; Andersson, M. Potent adjuvant effect by anti-CD40 in collagen-induced arthritis. Enhanced disease is accompanied by increased production of collagen type-II reactive IgG2a and IFN-gamma. J. Autoimmun. 2000, 14, 295–302. [Google Scholar] [CrossRef]
- Choi, E.W.; Lee, K.W.; Park, H.; Kim, H.; Lee, J.H.; Song, J.W.; Yang, J.; Kwon, Y.; Kim, T.M.; Park, J.B.; et al. Therapeutic effects of anti-CD154 antibody in cynomolgus monkeys with advanced rheumatoid arthritis. Sci. Rep. 2018, 8, 2135. [Google Scholar] [CrossRef]
- Ferrer, I.R.; Liu, D.; Pinelli, D.F.; Koehn, B.H.; Stempora, L.L.; Ford, M.L. CD40/CD154 blockade inhibits dendritic cell expression of inflammatory cytokines but not costimulatory molecules. J. Immunol. 2012, 189, 4387–4395. [Google Scholar] [CrossRef]
- Ferrer, I.R.; Wagener, M.E.; Song, M.; Kirk, A.D.; Larsen, C.P.; Ford, M.L. Antigen-specific induced Foxp3+ regulatory T cells are generated following CD40/CD154 blockade. Proc. Natl. Acad. Sci. USA 2011, 108, 20701–20706. [Google Scholar] [CrossRef]
- Warren, K.J.; Iwami, D.; Harris, D.G.; Bromberg, J.S.; Burrell, B.E. Laminins affect T cell trafficking and allograft fate. J. Clin. Invest. 2014, 124, 2204–2218. [Google Scholar] [CrossRef] [Green Version]
- Roser-Page, S.; Vikulina, T.; Yu, K.; McGee-Lawrence, M.E.; Weitzmann, M.N. Neutralization of CD40 ligand costimulation promotes bone formation and accretion of vertebral bone mass in mice. Rheumatology 2018, 57, 1105–1114. [Google Scholar] [CrossRef] [Green Version]
- Kronbichler, A.; Brezina, B.; Gauckler, P.; Quintana, L.F.; Jayne, D.R.W. Refractory lupus nephritis: When, why and how to treat. Autoimmun. Rev. 2019, 18, 510–518. [Google Scholar] [CrossRef]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef]
- Davis, J.C., Jr.; Totoritis, M.C.; Rosenberg, J.; Sklenar, T.A.; Wofsy, D. Phase I clinical trial of a monoclonal antibody against CD40-ligand (IDEC-131) in patients with systemic lupus erythematosus. J. Rheumatol. 2001, 28, 95–101. [Google Scholar]
- Kalunian, K.C.; Davis, J.C., Jr.; Merrill, J.T.; Totoritis, M.C.; Wofsy, D.; Group, I.-L.S. Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 3251–3258. [Google Scholar] [CrossRef]
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A.; Group, B.G.L.N.T. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727. [Google Scholar] [CrossRef]
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C.; Johnston, G.I.; Otoul, C.; Stach, C.; Zamacona, M.; Dorner, T.; Urowitz, M.; et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2017, 76, 1837–1844. [Google Scholar] [CrossRef]
- Correction: Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2018, 77, 787–788.[Green Version]
- Biogen Press Release: UCB and Biogen Announce Topline Results from a Phase 2b Study of Dapirolizumab Pegol in Systemic Lupus Erythematosus. 2018. Available online: http://investors.biogen.com/news-releases/news-release-details/ucb-and-biogen-announce-topline-results-phase-2b-study (accessed on 23 October 2018).
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11, eaar6584. [Google Scholar] [CrossRef]
- Gueiros, L.A.; France, K.; Posey, R.; Mays, J.W.; Carey, B.; Sollecito, T.P.; Setterfield, J.; Woo, S.B.; Culton, D.; Payne, A.S.; et al. World Workshop on Oral Medicine VII: Immunobiologics for salivary gland disease in Sjogren’s syndrome: A systematic review. Oral. Dis. 2019, 25, 102–110. [Google Scholar] [CrossRef]
- Visvanathan, S.; Daniluk, S.; Ptaszynski, R.; Muller-Ladner, U.; Ramanujam, M.; Rosenstock, B.; Eleftheraki, A.G.; Vinisko, R.; Petrikova, A.; Kellner, H.; et al. Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase IIa study. Ann. Rheum. Dis. 2019, 78, 754–760. [Google Scholar] [CrossRef]
- Liossis, S.N.; Sfikakis, P.P. Costimulation blockade in the treatment of rheumatic diseases. BioDrugs 2004, 18, 95–102. [Google Scholar] [CrossRef]
- Nakamura, M.; Tanaka, Y.; Satoh, T.; Kawai, M.; Hirakata, M.; Kaburaki, J.; Kawakami, Y.; Ikeda, Y.; Kuwana, M. Autoantibody to CD40 ligand in systemic lupus erythematosus: Association with thrombocytopenia but not thromboembolism. Rheumatology 2006, 45, 150–156. [Google Scholar] [CrossRef]
- Ferrant, J.L.; Benjamin, C.D.; Cutler, A.H.; Kalled, S.L.; Hsu, Y.M.; Garber, E.A.; Hess, D.M.; Shapiro, R.I.; Kenyon, N.S.; Harlan, D.M.; et al. The contribution of Fc effector mechanisms in the efficacy of anti-CD154 immunotherapy depends on the nature of the immune challenge. Int. Immunol. 2004, 16, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.H.; Yamniuk, A.P.; Borowski, V.; Kuhn, R.; Susulic, V.; Rex-Rabe, S.; Yang, X.; Zhou, X.; Zhang, Y.; Gillooly, K.; et al. Engineering of a novel anti-CD40L domain antibody for treatment of autoimmune diseases. J. Immunol. 2014, 192, 4083–4092. [Google Scholar] [CrossRef]
- Shock, A.; Burkly, L.; Wakefield, I.; Peters, C.; Garber, E.; Ferrant, J.; Taylor, F.R.; Su, L.; Hsu, Y.M.; Hutto, D.; et al. CDP7657, an anti-CD40L antibody lacking an Fc domain, inhibits CD40L-dependent immune responses without thrombotic complications: An in vivo study. Arthritis Res. Ther. 2015, 17, 234. [Google Scholar] [CrossRef]
- Albach, F.N.; Wagner, F.; Huser, A.; Igel, J.; Joseph, D.; Hilbert, J.; Schoelch, C.; Padula, S.J.; Steffgen, J. Safety, pharmacokinetics and pharmacodynamics of single rising doses of BI 655064, an antagonistic anti-CD40 antibody in healthy subjects: A potential novel treatment for autoimmune diseases. Eur. J. Clin. Pharm. 2018, 74, 161–169. [Google Scholar] [CrossRef]
- Schwabe, C.; Rosenstock, B.; Doan, T.; Hamilton, P.; Dunbar, P.R.; Eleftheraki, A.G.; Joseph, D.; Hilbert, J.; Schoelch, C.; Padula, S.J.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of Multiple Rising Doses of BI 655064, an Antagonistic Anti-CD40 Antibody, in Healthy Subjects: A Potential Novel Treatment for Autoimmune Diseases. J. Clin. Pharm. 2018, 58, 1566–1577. [Google Scholar] [CrossRef]
- Lai, J.H.; Ling, X.C.; Ho, L.J. Useful message in choosing optimal biological agents for patients with autoimmune arthritis. Biochem. Pharm. 2019, 165, 99–111. [Google Scholar] [CrossRef]
- Lee, J.I.; Choi, Y.J.; Park, H.J.; Jung, K.C.; Park, S.H. RD-05, a novel anti-CD154 antibody, efficiently inhibits generation of anti-drug antibody without the risk of thrombus formation in non-human primates. Biochem. Biophys. Res. Commun. 2018, 498, 996–1001. [Google Scholar] [CrossRef]
- Thompson, P.; Cardona, K.; Russell, M.; Badell, I.R.; Shaffer, V.; Korbutt, G.; Rayat, G.R.; Cano, J.; Song, M.; Jiang, W.; et al. CD40-specific costimulation blockade enhances neonatal porcine islet survival in nonhuman primates. Am. J. Transpl. 2011, 11, 947–957. [Google Scholar] [CrossRef]
- Silvian, L.F.; Friedman, J.E.; Strauch, K.; Cachero, T.G.; Day, E.S.; Qian, F.; Cunningham, B.; Fung, A.; Sun, L.; Shipps, G.W.; et al. Small molecule inhibition of the TNF family cytokine CD40 ligand through a subunit fracture mechanism. ACS Chem. Biol. 2011, 6, 636–647. [Google Scholar] [CrossRef]
- Anil Kumar, M.S.; Papp, K.; Tainaka, R.; Valluri, U.; Wang, X.; Zhu, T.; Schwabe, C. Randomized, controlled study of bleselumab (ASKP1240) pharmacokinetics and safety in patients with moderate-to-severe plaque psoriasis. Biopharm. Drug. Dispos. 2018, 39, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Ma, A.; Dun, H.; Hu, Y.; Zeng, L.; Bai, J.; Zhang, G.; Kinugasa, F.; Sudo, Y.; Miyao, Y.; et al. Effects of ASKP1240 combined with tacrolimus or mycophenolate mofetil on renal allograft survival in Cynomolgus monkeys. Transplantation 2014, 98, 267–276. [Google Scholar] [CrossRef]
- Watanabe, M.; Yamashita, K.; Suzuki, T.; Kamachi, H.; Kuraya, D.; Koshizuka, Y.; Ogura, M.; Yoshida, T.; Aoyagi, T.; Fukumori, D.; et al. ASKP1240, a fully human anti-CD40 monoclonal antibody, prolongs pancreatic islet allograft survival in nonhuman primates. Am. J. Transpl. 2013, 13, 1976–1988. [Google Scholar] [CrossRef]
- Allen, R.C.; Armitage, R.J.; Conley, M.E.; Rosenblatt, H.; Jenkins, N.A.; Copeland, N.G.; Bedell, M.A.; Edelhoff, S.; Disteche, C.M.; Simoneaux, D.K.; et al. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science 1993, 259, 990–993. [Google Scholar] [CrossRef]
- DiSanto, J.P.; Bonnefoy, J.Y.; Gauchat, J.F.; Fischer, A.; de Saint Basile, G. CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature 1993, 361, 541–543. [Google Scholar] [CrossRef]
- Aruffo, A.; Farrington, M.; Hollenbaugh, D.; Li, X.; Milatovich, A.; Nonoyama, S.; Bajorath, J.; Grosmaire, L.S.; Stenkamp, R.; Neubauer, M.; et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell 1993, 72, 291–300. [Google Scholar] [CrossRef]
- Xu, J.; Foy, T.M.; Laman, J.D.; Elliott, E.A.; Dunn, J.J.; Waldschmidt, T.J.; Elsemore, J.; Noelle, R.J.; Flavell, R.A. Mice deficient for the CD40 ligand. Immunity 1994, 1, 423–431. [Google Scholar] [CrossRef]
- Renshaw, B.R.; Fanslow, W.C., 3rd; Armitage, R.J.; Campbell, K.A.; Liggitt, D.; Wright, B.; Davison, B.L.; Maliszewski, C.R. Humoral immune responses in CD40 ligand-deficient mice. J. Exp. Med. 1994, 180, 1889–1900. [Google Scholar] [CrossRef]
- Ferrari, S.; Giliani, S.; Insalaco, A.; Al-Ghonaium, A.; Soresina, A.R.; Loubser, M.; Avanzini, M.A.; Marconi, M.; Badolato, R.; Ugazio, A.G.; et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natl. Acad. Sci. USA 2001, 98, 12614–12619. [Google Scholar] [CrossRef]
- Revy, P.; Muto, T.; Levy, Y.; Geissmann, F.; Plebani, A.; Sanal, O.; Catalan, N.; Forveille, M.; Dufourcq-Labelouse, R.; Gennery, A.; et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 2000, 102, 565–575. [Google Scholar] [CrossRef]
- Imai, K.; Slupphaug, G.; Lee, W.I.; Revy, P.; Nonoyama, S.; Catalan, N.; Yel, L.; Forveille, M.; Kavli, B.; Krokan, H.E.; et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003, 4, 1023–1028. [Google Scholar] [CrossRef]
- Beatty, G.L.; Li, Y.; Long, K.B. Cancer immunotherapy: Activating innate and adaptive immunity through CD40 agonists. Expert. Rev. Anticancer Ther. 2017, 17, 175–186. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Aimed Target | Drugs | Structure | Aimed Diseases | Trial Designs | Patient No./Study Period | Results and Comments | Ref. |
|---|---|---|---|---|---|---|---|
| CD154 | IDEC-131 | Humanized mAb | SLE * | Phase I & Phase II, DB, PC | 23/3 months and 85/7 months | Safe and well-tolerated and no clinical benefits compared to baseline | [91,92] |
| CD154 | BG9588 | Humanized mAb | SLE (PLN) | phase II, open-label | 28/22–24 wks | Early termination of the trial due to thromboembolic AE although clinical benefits noted | [93] |
| CD154 | Dapirolizumab pegol | Fab | SLE | Phase I, DB & Phase IIb, DB, PC | 16/12 wks and 182/24 wks | Clinical benefits in high disease activity group at baseline & Fail to meet the primary endpoint (noted in press release) | [94,95,96] |
| CD154 | VIB4920 (MEDI4920) | Tn3 fusion protein | RA | Phase Ib, DB, PC | 57/12 wks | Significant benefits in clinical and laboratorial assessment | [97] |
| CD40 | CFZ533 | Humanized mAb | SS; RA | Phase IIa, DB, PC & Phase I, DB, PC | 44/12 wks and 75/20 wks (estimated) | Clinical benefits in SS patients & The drug was well-tolerated in RA patients | [18,98], NCT02-089087 |
| CD40 | BI 655064 | Humanized mAb | RA, fail with MTX | Phase IIa, DB, PC | 67/12 wks | Improvement in laboratorial parameters but no significant clinical efficacy | [99] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, J.-H.; Luo, S.-F.; Ho, L.-J. Targeting the CD40-CD154 Signaling Pathway for Treatment of Autoimmune Arthritis. Cells 2019, 8, 927. https://doi.org/10.3390/cells8080927
Lai J-H, Luo S-F, Ho L-J. Targeting the CD40-CD154 Signaling Pathway for Treatment of Autoimmune Arthritis. Cells. 2019; 8(8):927. https://doi.org/10.3390/cells8080927
Chicago/Turabian StyleLai, Jenn-Haung, Shue-Fen Luo, and Ling-Jun Ho. 2019. "Targeting the CD40-CD154 Signaling Pathway for Treatment of Autoimmune Arthritis" Cells 8, no. 8: 927. https://doi.org/10.3390/cells8080927