
Post-Translational Modifications of RelB NF-κB Subunit and Associated Functions


Abstract
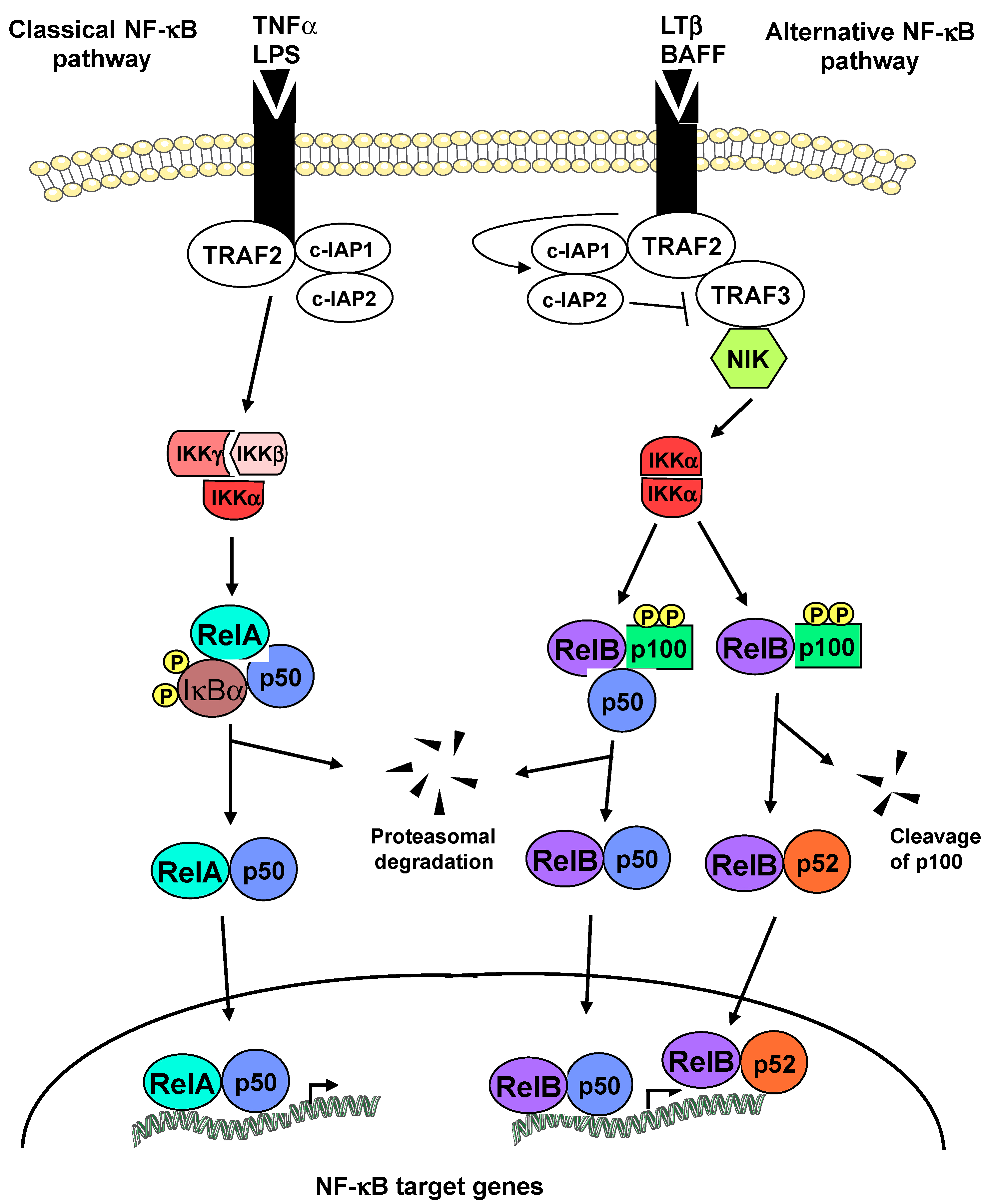
:1. Introduction
2. Phosphorylation of RelB
2.1. Serine 552 and Threonine 84
2.2. Serine 368
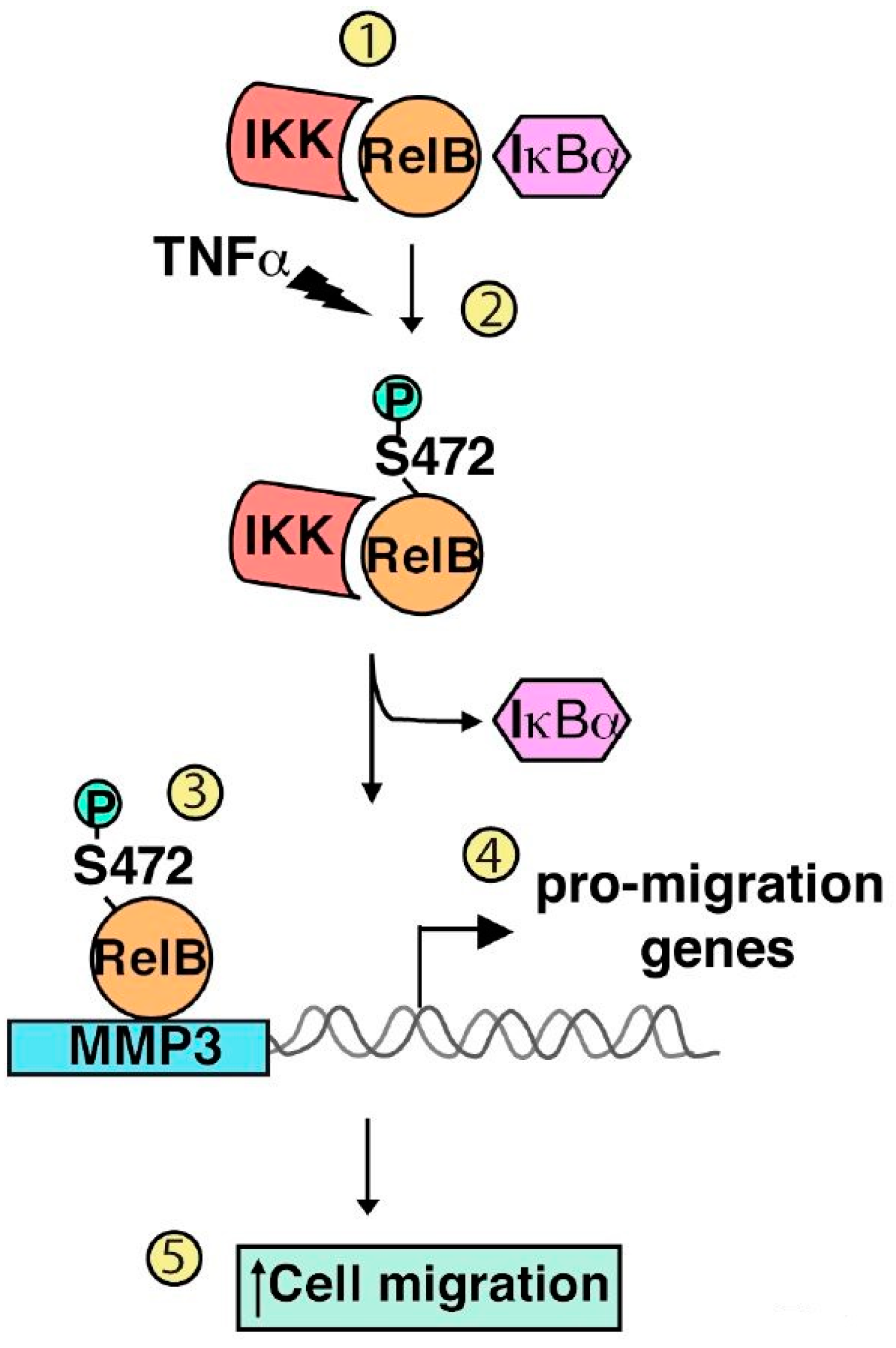
2.3. Serine 472
2.4. Other Putative Phosphorylation Sites
3. Polyubiquitination of RelB
4. SUMOylation of RelB
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-κB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Karin, M. Nuclear factor-κB—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [PubMed]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-κB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Rayet, B.; Gélinas, C. Aberrant Rel/NF-κB genes and activity in human cancer. Oncogene 1999, 18, 6938–6947. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. NF-κB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, C.; Toi, M. Nuclear factor factor-κB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Jacque, E. The alternative NF-κB activation pathway and cancer: Friend or foe? Med. Sci. (Paris) 2008, 24, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Azarenko, V.; Dejardinn, E.; Siebenlist, U. Human RelB (I-Rel) functions as a κ B site-dependent transactivating member of the family of Rel-related proteins. Oncogene 1994, 9, 1699–1702. [Google Scholar] [PubMed]
- Bours, V.; Burd, P.R.; Brown, K.; Villalobos, J.; Park, S.; Ryseck, R.P.; Bravo, R.; Kelly, K.; Siebenlist, U. A novel mitogen-inducible gene product related to p50/p105-NF-κ B participates in transactivation through a κB site. Mol. Cell Biol. 1992, 12, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Ryseck, R.P.; Bull, P.; Takamiya, M.; Bours, V.; Siebenlist, U.; Dobrzanski, P.; Bravo, R. RelB, a new Rel family transcription activator that can interact with p50-NF-κB. Mol. Cell Biol. 1992, 12, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Both N- and C-terminal domains of RelB are required for full transactivation: Role of the N-terminal leucine zipper-like motif. Mol. Cell Biol. 1993, 13, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Tchenio, T.; Piton, G.; Romeo, P.H.; Baud, V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 14635–14640. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.B.; Weih, D.S.; Sivakumar, V.; Weih, F. RelB is required for Peyer’s patch development: Differential regulation of p52-RelB by lymphotoxin and TNF. Embo J. 2003, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Korner, M.; Raud, V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation: Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. Embo J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.J.; Huang, D.B.; Miller, D.; Wang, V.Y.; Vu, D.; Ghosh, G. NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes. EMBO Rep. 2009, 10, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; De Santa, F. Shaping alternative NF-κB-dependent gene expression programs: New clues to specificity. Cell Death Differ. 2006, 13, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. Modulation of NF-κB activity by exchange of dimers. Mol. Cell 2003, 11, 1563–1574. [Google Scholar] [CrossRef]
- Wu, L.; D'Amico, A.; Winkel, K.D.; Suter, M.; Lo, D.; Shortman, K. RelB is essential for the development of myeloid-related CD8α-dendritic cells but not of lymphoid-related CD8α+ dendritic cells. Immunity 1998, 9, 839–847. [Google Scholar] [CrossRef]
- Weih, F.; Carrasco, D.; Durham, S.K.; Barton, D.S.; Rizzo, C.A.; Ryseck, R.P.; Lira, S.A.; Bravo, R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κ B/Rel family. Cell 1995, 80, 331–340. [Google Scholar] [CrossRef]
- Weih, F.; Warr, G.; Yang, H.; Bravo, R. Multifocal defects in immune responses in RelB-deficient mice. J. Immunol. 1997, 158, 5211–5218. [Google Scholar] [PubMed]
- Weih, D.S.; Yilmaz, Z.B.; Weih, F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J. Immunol. 2001, 167, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Weih, F.; Durham, S.K.; Barton, D.S.; Sha, W.C.; Baltimore, D.; Bravo, R. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent. J. Immunol. 1996, 157, 3974–3979. [Google Scholar] [PubMed]
- Zhu, H.C.; Qiu, T.; Liu, X.H.; Dong, W.C.; Weng, X.D.; Hu, C.H.; Kuang, Y.L.; Gao, R.H.; Dan, C.; Tao, T. Tolerogenic dendritic cells generated by RelB silencing using shRNA prevent acute rejection. Cell Immunol. 2012, 274, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Wu, D.; Goth, S.R.; Baek, J.; Lollies, A.; Domhardt, R.; Grindel, A.; Pessah, I.N. Aryl hydrocarbon receptor signaling regulates NF-κB RelB activation during dendritic-cell differentiation. Immunol. Cell Biol. 2013, 91, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Yoza, B.K.; Hu, J.Y.; Cousart, S.L.; Forrest, L.M.; McCall, C.E. Induction of RelB participates in endotoxin tolerance. J. Immunol. 2006, 177, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M.; Yoza, B.K.; Hu, J.Y.Q.; Cousart, S.L.; McCall, C.E. Epigenetic Silencing of Tumor Necrosis Factor α during Endotoxin Tolerance. J. Biol. Chem. 2007, 282, 26857–26864. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Pauza, M.E.; Feng, L.; Lo, D. RelB regulation of chemokine expression modulates local inflammation. Am. J. Pathol. 1997, 151, 375–387. [Google Scholar] [PubMed]
- Xia, Y.; Chen, S.; Wang, Y.; Mackman, N.; Ku, G.; Lo, D.; Feng, L. RelB modulation of IκBα stability as a mechanism of transcription suppression of interleukin-1α (IL-1α), IL-1β, and tumor necrosis factor α in fibroblasts. Mol. Cell Biol. 1999, 19, 7688–7696. [Google Scholar] [CrossRef] [PubMed]
- Bakkar, N.; Ladner, K.; Canan, B.D.; Liyanarachchi, S.; Bal, N.C.; Pant, M.; Periasamy, M.; Li, Q.; Janssen, P.M.; Guttridge, D.C. IKKα and alternative NF-κB regulate PGC-1β to promote oxidative muscle metabolism. J. Cell Biol. 2012, 196, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [PubMed]
- Millet, P.; McCall, C.; Yoza, B. RelB: An outlier in leukocyte biology. J. Leukoc. Biol. 2013, 94, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Bellet, M.M.; Zocchi, L.; Sassone-Corsi, P. The RelB subunit of NFkB acts as a negative regulator of circadian gene expression. Cell Cycle 2012, 11, 3304–3311. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–3955. [Google Scholar] [CrossRef] [PubMed]
- Baglole, C.J.; Maggirwar, S.B.; Gasiewicz, T.A.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-κB family member RelB. J. Biol. Chem. 2008, 283, 28944–28957. [Google Scholar] [CrossRef] [PubMed]
- Vaira, S.; Johnson, T.; Hirbe, A.C.; Alhawagri, M.; Anwisye, I.; Sammut, B.; O’Neal, J.; Zou, W.; Weilbaecher, K.N.; Faccio, R.; et al. RelB is the NF-κB subunit downstream of NIK responsible for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Soysa, N.S.; Alles, N.; Weih, D.; Lovas, A.; Mian, A.H.; Shimokawa, H.; Yasuda, H.; Weih, F.; Jimi, E.; Ohya, K.; et al. The pivotal role of the alternative NF-κB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J. Bone Miner. Res. 2010, 25, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, R.; Fukushima, H.; Osawa, K.; Maruyama, T.; Yasuda, H.; Weih, F.; Doi, T.; Maki, K.; Jimi, E. RelB-induced expression of Cot, an MAP3K family member, rescues RANKL-induced osteoclastogenesis in alymphoplasia mice by promoting NF-κB2 processing by IKKα. J. Biol. Chem. 2014, 289, 7349–7361. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, A.; Chaurushiya, M.; Singh, B.; Levine, A.J. Activation of NF-κB and inhibition of p53-mediated apoptosis by API2/mucosa-associated lymphoid tissue 1 fusions promote oncogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 9079–9084. [Google Scholar] [CrossRef] [PubMed]
- Cormier, F.; Monjanel, H.; Fabre, C.; Billot, K.; Sapharikas, E.; Chereau, F.; Bordereaux, D.; Molina, T.J.; Avet-Loiseau, H.; Baud, V. Frequent engagement of RelB activation is critical for cell survival in multiple myeloma. PLoS ONE 2013, 8, e59127. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Dorken, B.; Jundt, F. Notch is an essential upstream regulator of NF-κB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia 2012, 26, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Lwin, T.; Hazlehurst, L.A.; Li, Z.; Dessureault, S.; Sotomayor, E.; Moscinski, L.C.; Dalton, W.S.; Tao, J. Bone marrow stromal cells prevent apoptosis of lymphoma cells by up regulation of anti-apoptotic proteins associated with activation of NF-κB (RelB/p52) in non-Hodgkin's lymphoma cells. Leukemia 2007, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.R.; Williame, M.; Gachet, S.; Cormier, F.; Janin, A.; Weih, D.; Weih, F.; Ghysdael, J. RelB-dependent stromal cells promote T-cell leukemogenesis. PLoS ONE 2008, 3, e2555. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Josson, S.; Fang, F.; Oberley, T.D.; St Clair, D.K.; Wan, X.S.; Sun, Y.; Bakthavatchalu, V.; Muthuswamy, A.; St Clair, W.H. RelB enhances prostate cancer growth: Implications for the role of the nuclear factor-κB alternative pathway in tumorigenicity. Cancer Res. 2009, 69, 3267–3271. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Ramakrishnan, D.; Valenta, J.; Parney, I.F.; Bayless, K.J.; Sitcheran, R. The NF-κB RelB protein is an oncogenic driver of mesenchymal glioma. PLoS ONE 2013, 8, e57489. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Duan, X.; Zhou, P.; Zhou, W.; Wu, X.; Xu, S.; Chen, Y.; Tao, Z. Lymphotoxin β receptor activation promotes bladder cancer in a nuclear factor-κB-dependent manner. Mol. Med. Rep. 2015, 11, 783–790. [Google Scholar] [PubMed]
- Dimitrakopoulos, F.I.; Antonacopoulou, A.G.; Kottorou, A.; Vlotinou, H.; Panagopoulos, N.D.; Dougenis, D.; Scopa, C.; Papadaki, H.; Kalofonos, H.P. NSCLC and the alternative pathway of NF-κB: Uncovering an unknown relation. Virchows Arch. 2012, 460, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Josson, S.; Xu, Y.; Fang, F.; Dhar, S.K.; St Clair, D.K.; St Clair, W.H. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene 2006, 25, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, F.; St Clair, D.K.; Josson, S.; Sompol, P.; Spasojevic, I.; St Clair, W.H. Suppression of RelB-mediated manganese superoxide dismutase expression reveals a primary mechanism for radiosensitization effect of 1α,25-dihydroxyvitamin D(3) in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Cherry, E.M.; Lee, D.W.; Jung, J.U.; Sitcheran, R. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioma cell invasion through induction of NF-κB-inducing kinase (NIK) and noncanonical NF-κB signaling. Mol. Cancer 2015, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Demicco, E.G.; Kavanagh, K.T.; Romieu-Mourez, R.; Wang, X.; Shin, S.R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. RelB/p52 NF-κB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IκB-α expression and promote carcinogenesis of the mammary gland. Mol. Cell Biol. 2005, 25, 10136–10147. [Google Scholar] [CrossRef] [PubMed]
- Mineva, N.D.; Wang, X.; Yang, S.; Ying, H.; Xiao, Z.X.; Holick, M.F.; Sonenshein, G.E. Inhibition of RelB by 1,25-dihydroxyvitamin D3 promotes sensitivity of breast cancer cells to radiation. J. Cell Physiol. 2009, 220, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene 2006, 25, 6717–6830. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-κB: Another layer of regulation for NF-κB signaling pathway. Cell Signal 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Calao, M.; Burny, A.; Quivy, V.; Dekoninck, A.; Van Lint, C. A pervasive role of histone acetyltransferases and deacetylases in an NF-κB-signaling code. Trends Biochem. Sci. 2008, 33, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Stark, G.R. NF-κB: Regulation by Methylation. Cancer Res. 2015, 75, 3692–3695. [Google Scholar] [CrossRef] [PubMed]
- Schmukle, A.C.; Walczak, H. No one can whistle a symphony alone—How different ubiquitin linkages cooperate to orchestrate NF-κB activity. J. Cell Sci. 2012, 125, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Iwai, K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol. Rev. 2015, 266, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabb, A.M.; Miyamoto, S. SUMO and NF-κB ties. Cell Mol. Life Sci. 2007, 64, 1979–1996. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Hierarchies of NF-κB target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Msaki, A.; Sánchez, A.M.; Koh, L.F.; Barré, B.; Rocha, S.; Perkins, N.D.; Johnson, R.F. The role of RelA (p65) threonine 505 phosphorylation in the regulation of cell growth, survival, and migration. Mol. Biol. Cell 2011, 22, 3032–3040. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yarbrough, W.G. Negative regulation of RelA phosphorylation: Emerging players and their roles in cancer. Cytokine Growth Factor Rev. 2015, 26, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Marienfeld, R.; Berberich-Siebelt, F.; Berberich, I.; Denk, A.; Serfling, E.; Neumann, M. Signal-specific and phosphorylation-dependent RelB degradation: A potential mechanism of NF-κB control. Oncogene 2001, 20, 8142–8147. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Marienfeld, R.; Wirth, T.; Baumann, B. Critical role of RelB serine 368 for dimerization and p100 stabilization. J. Biol. Chem. 2003, 278, 39242–39250. [Google Scholar] [CrossRef] [PubMed]
- Leidner, J.; Palkowitsch, L.; Marienfeld, U.; Fischer, D.; Marienfeld, R. Identification of lysine residues critical for the transcriptional activity and polyubiquitination of the NF-κB family member RelB. Biochem. J. 2008, 416, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Authier, H.; Billot, K.; Derudder, E.; Bordereaux, D.; Rivière, P.; Rodrigues-Ferreira, S.; Nahmias, C.; Baud, V. IKK phosphorylates RelB to modulate its promoter specificity and promote fibroblast migration downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 14794–14799. [Google Scholar] [CrossRef] [PubMed]
- Leidner, J.; Voogdt, C.; Niedenthal, R.; Möller, P.; Marienfeld, U.; Marienfeld, R.B. SUMOylation attenuates the transcriptional activity of the NF-κB subunit RelB. J. Cell Biochem. 2014, 115, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Hailfinger, S.; Nogai, H.; Pelzer, C.; Jaworski, M.; Cabalzar, K.; Charton, J.-E.; Guzzardi, M.; Décaillet, C.; Grau, M.; Dörken, B.; et al. Malt1-dependent RelB cleavage promotes canonical NF-κB activation in lymphocytes and lymphoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 14596–14601. [Google Scholar] [CrossRef] [PubMed]
- Phosphosite. Available online: http://www.phosphosite.org (accessed on 28 April 2016).
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Puto, L.A.; Reed, J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008, 22, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Gill, G. SUMO and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Dev. 2004, 18, 2046–2059. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Modification | Site(s) | Enzyme(s) | Effect | Reference |
|---|---|---|---|---|
| Phosphorylation | Threonine 84, Serine 552 | Unknown | Degradation | Marienfeld et al. 2001 [71] |
| Phosphorylation | Serine 368 | Unknown | Dimerization | Maier et al. 2003 [72] |
| Polyubiquitination | Lysine 273, 274, 305 and 308 | Unknown | Transcriptional activity | Leidner et al. 2008 [73] |
| Phosphorylation | Serine 472 | IKKα/IKKβ | Cell migration | Authier et al. 2014 [74] |
| SUMOylation | Lysine 387, 388, 390, 411, 414, 415, and 416 | Unknown | Transcriptional activity | Leidner et al. 2014 [75] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baud, V.; Collares, D. Post-Translational Modifications of RelB NF-κB Subunit and Associated Functions. Cells 2016, 5, 22. https://doi.org/10.3390/cells5020022
Baud V, Collares D. Post-Translational Modifications of RelB NF-κB Subunit and Associated Functions. Cells. 2016; 5(2):22. https://doi.org/10.3390/cells5020022
Chicago/Turabian StyleBaud, Véronique, and Davi Collares. 2016. "Post-Translational Modifications of RelB NF-κB Subunit and Associated Functions" Cells 5, no. 2: 22. https://doi.org/10.3390/cells5020022