

Functional and Multi-Omics Effects of an Optimized CRISPR-Mediated FURIN Depletion in U937 Monocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Plasmid Preparation
2.3. Cell Culture, Transfection and Cell Proliferation Assays
2.4. PCR Screening for CRISPR
2.5. Western Blot
2.6. Lipid Uptake Assay
2.7. Phagocytosis Assay
2.8. Migration Assay
2.9. Inflammatory Gene Expression via Quantitative PCR Assays
2.10. RNA Sequencing Analysis
2.11. Pathway Enrichment Analysis
2.12. Self-Organizing Map (SOM) Analysis
2.13. Targeted Proteomic Analysis
2.14. Data Analysis
3. Results
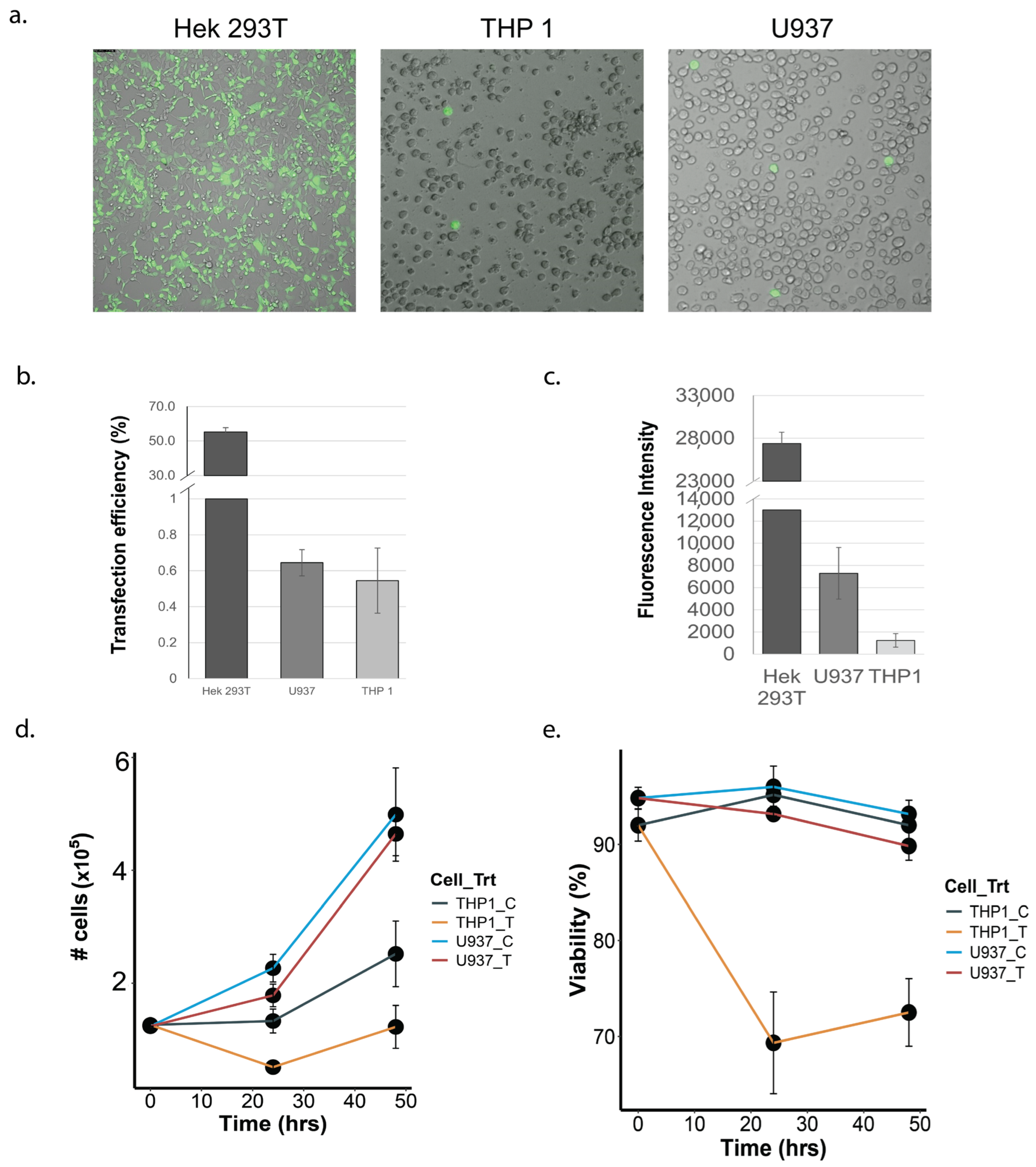
3.1. Transfection Efficiency and Post-Transfection Viability of THP-1 and U937 Monocytes
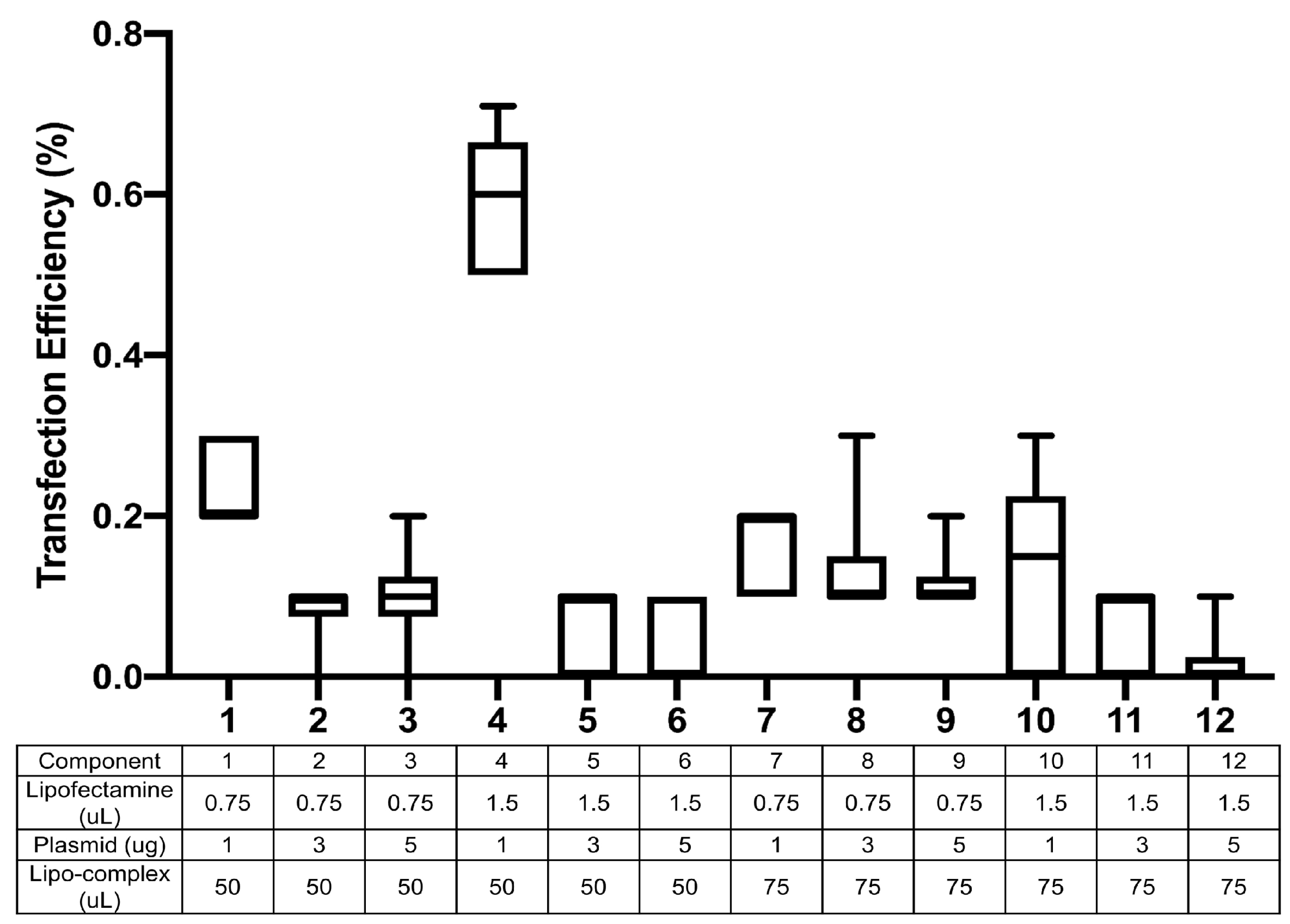
3.2. Transfection Optimization
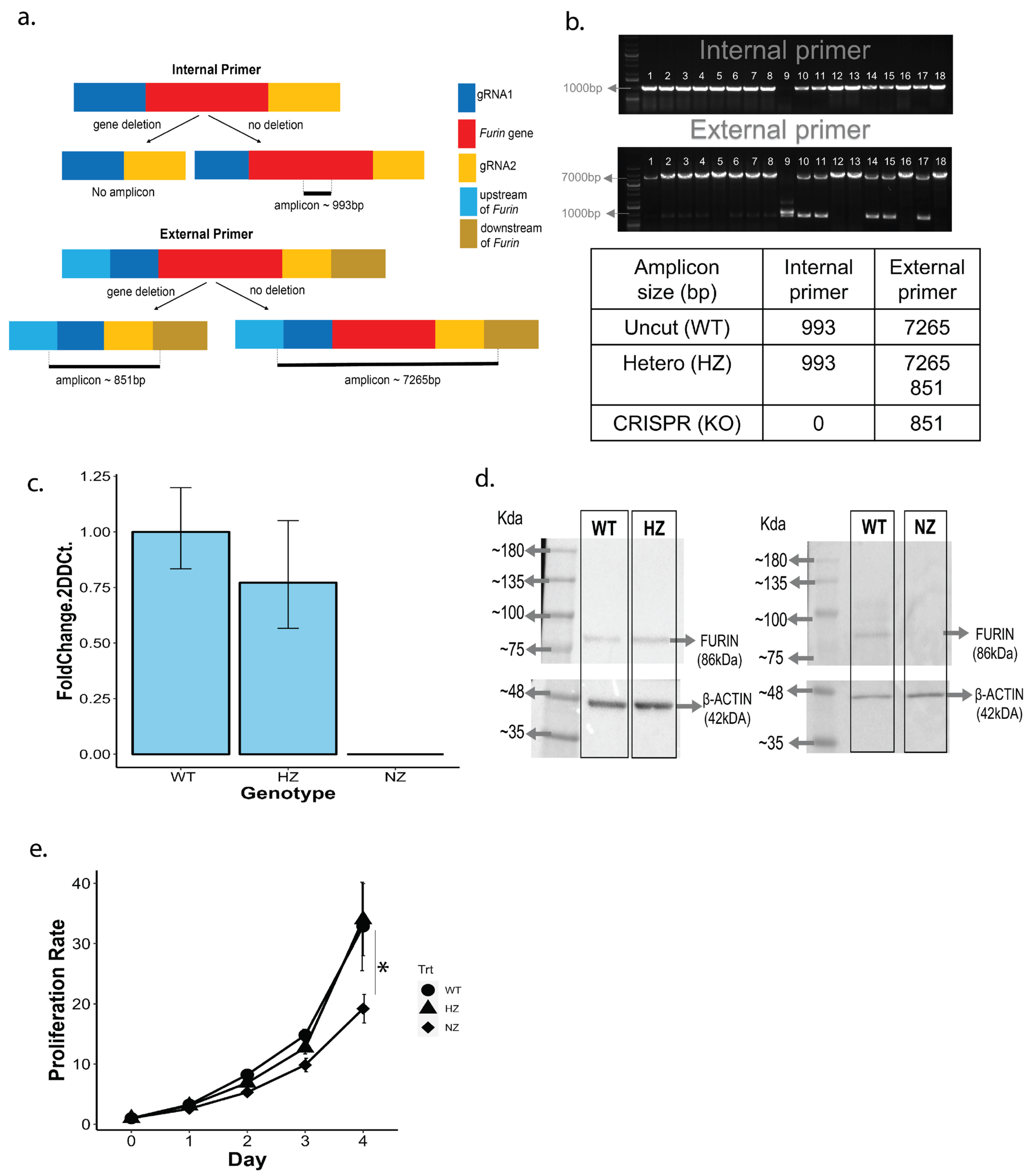
3.3. PCR Screening of CRISPR Candidate Clones
3.4. Validation of NZ and HZ Clones via qPCR and Protein Blotting
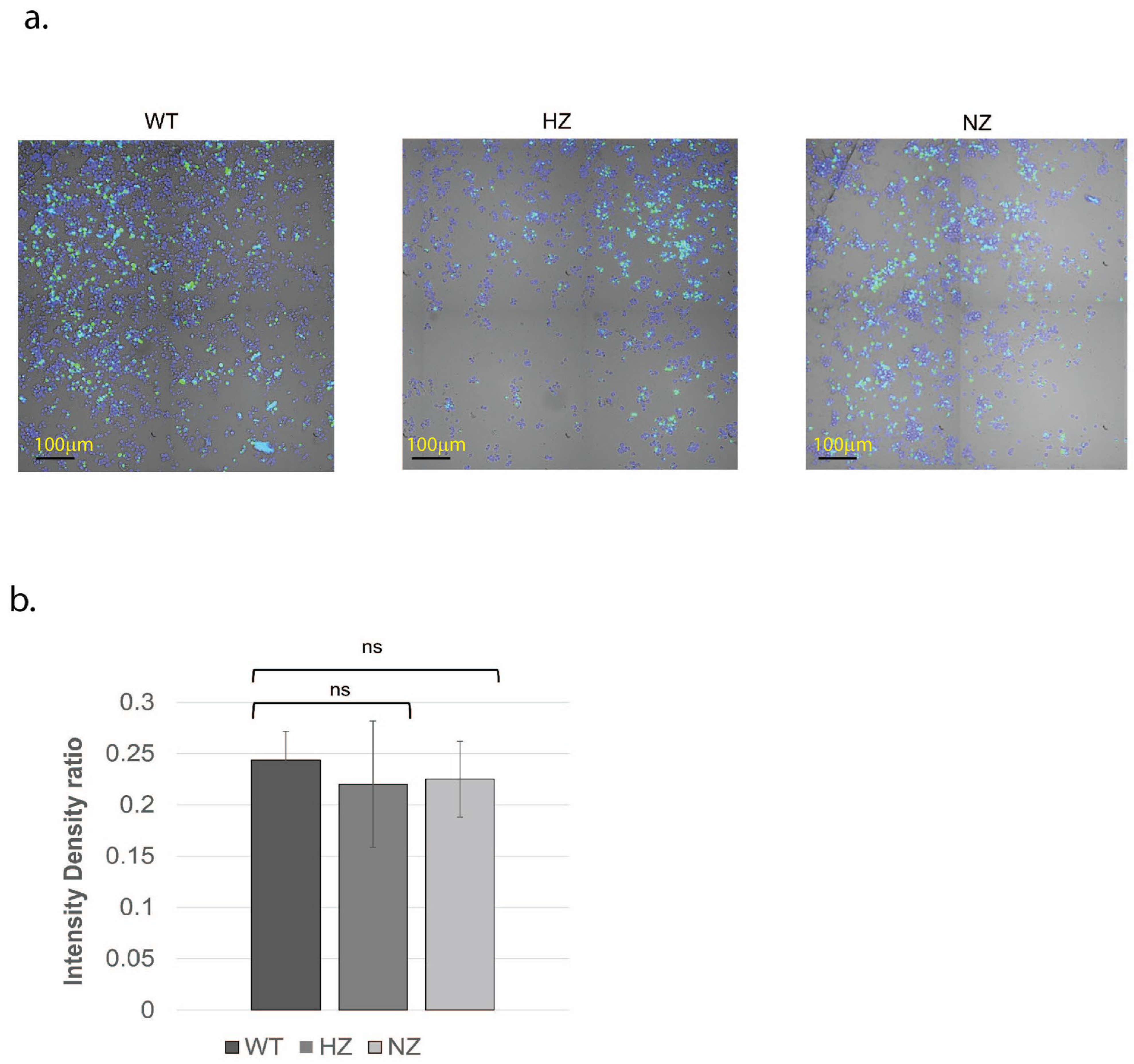
3.5. Loss of FURIN Does Not Modulate Macrophage Phagocytosis
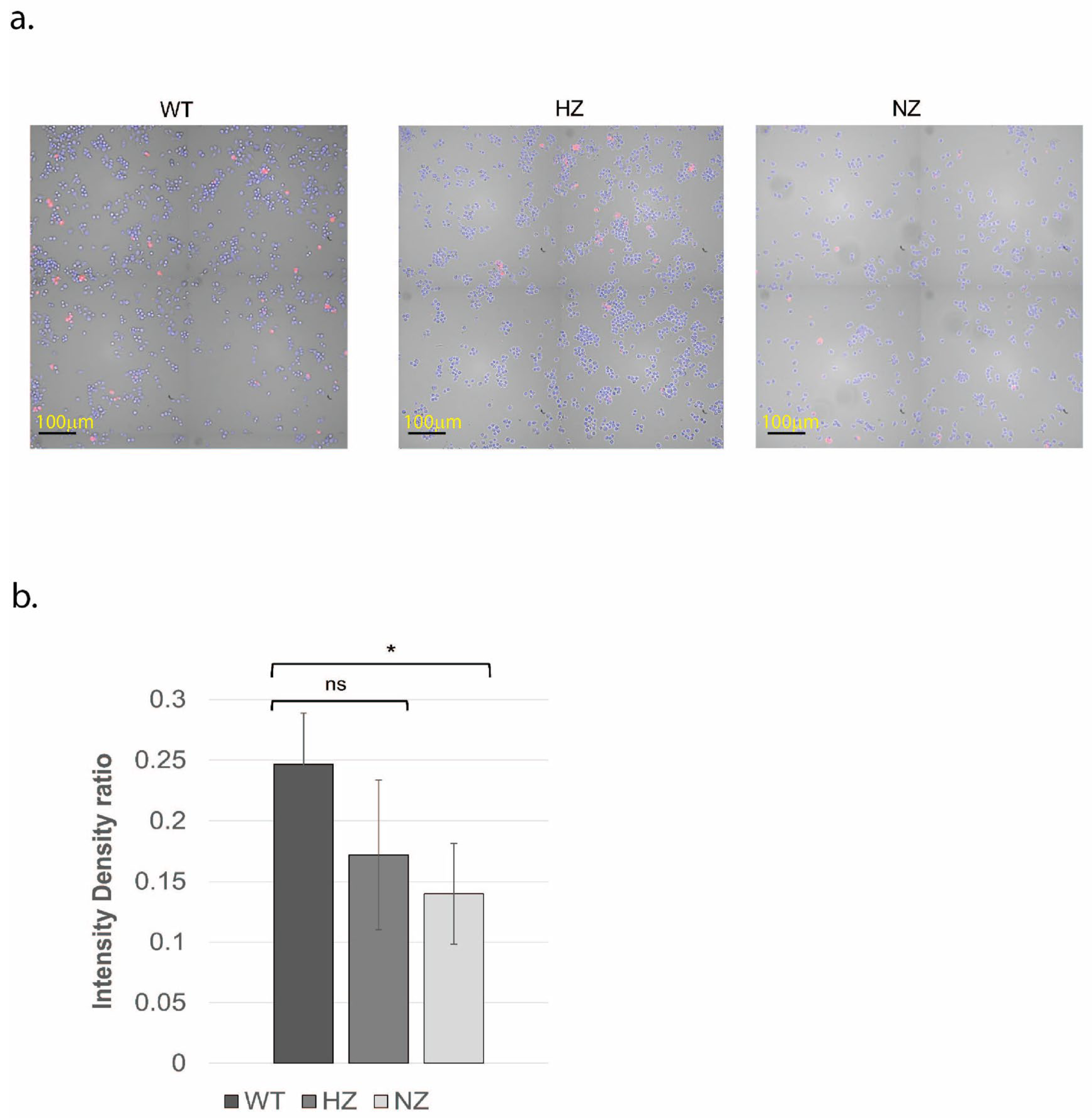
3.6. Loss of FURIN Reduces Macrophage LDL Accumulation
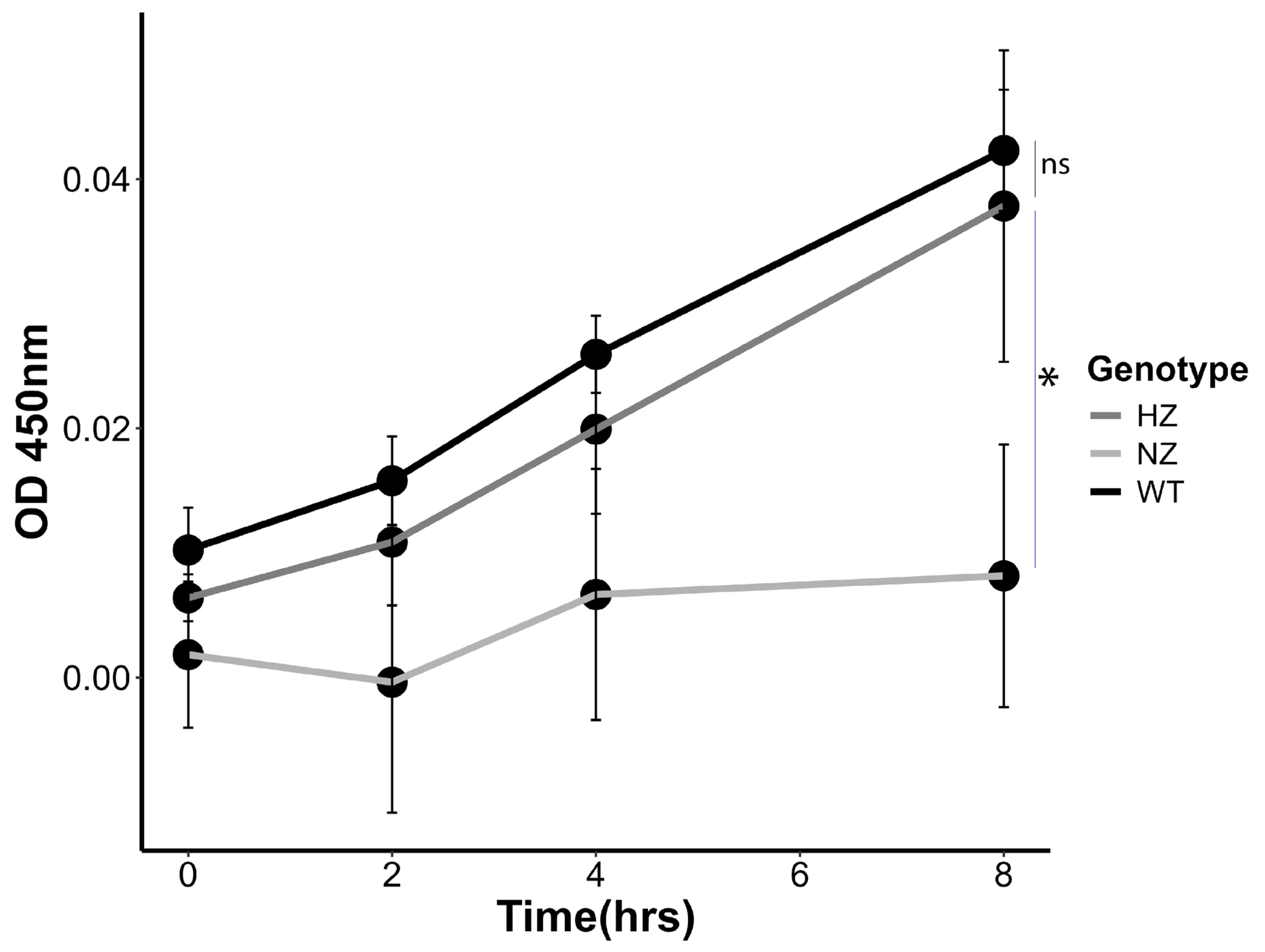
3.7. Loss of FURIN Reduces Monocyte Trans-Migration
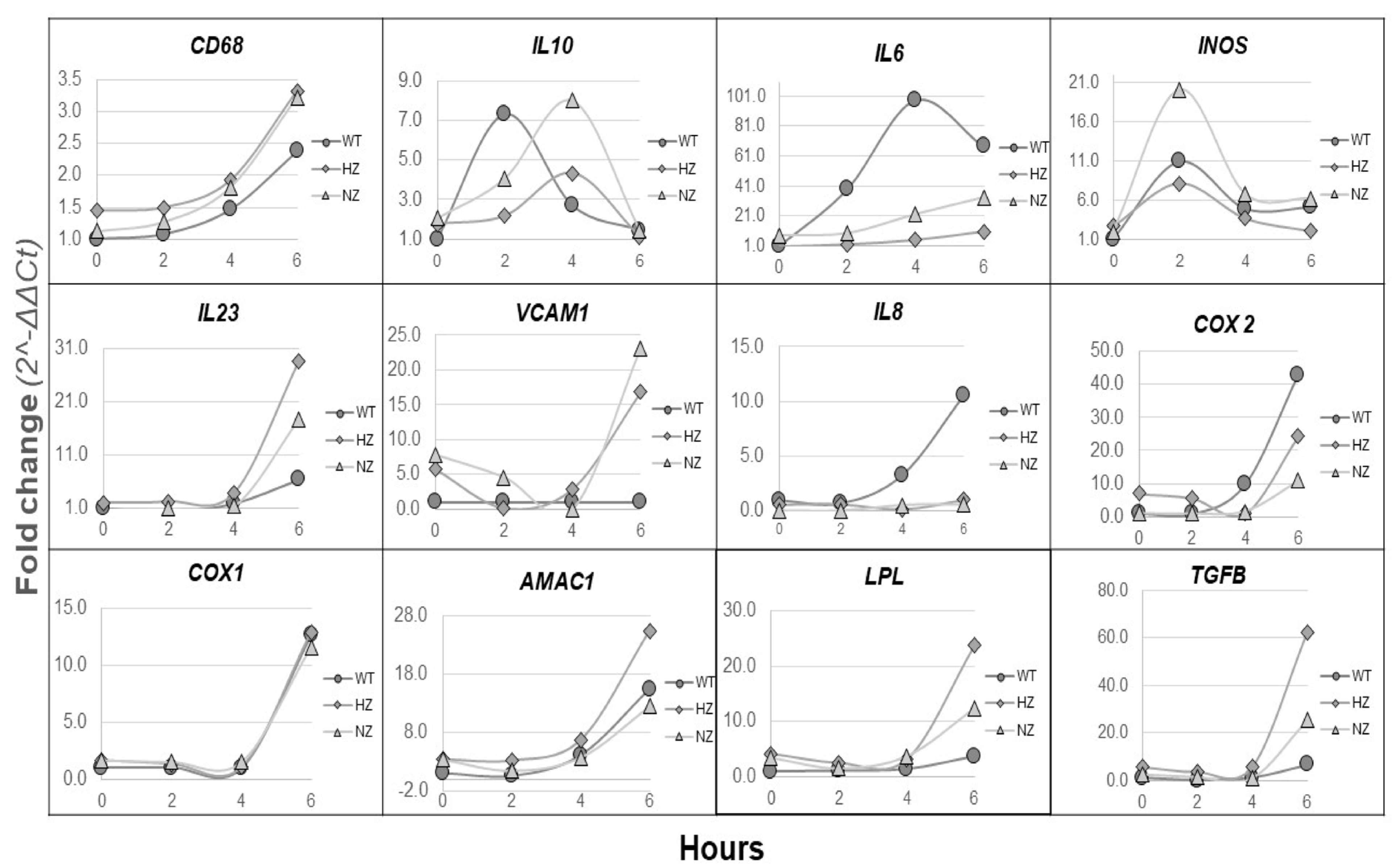
3.8. Loss of FURIN Shows Variable Effects on Inflammatory Gene Expression
3.9. RNA Sequencing Analysis
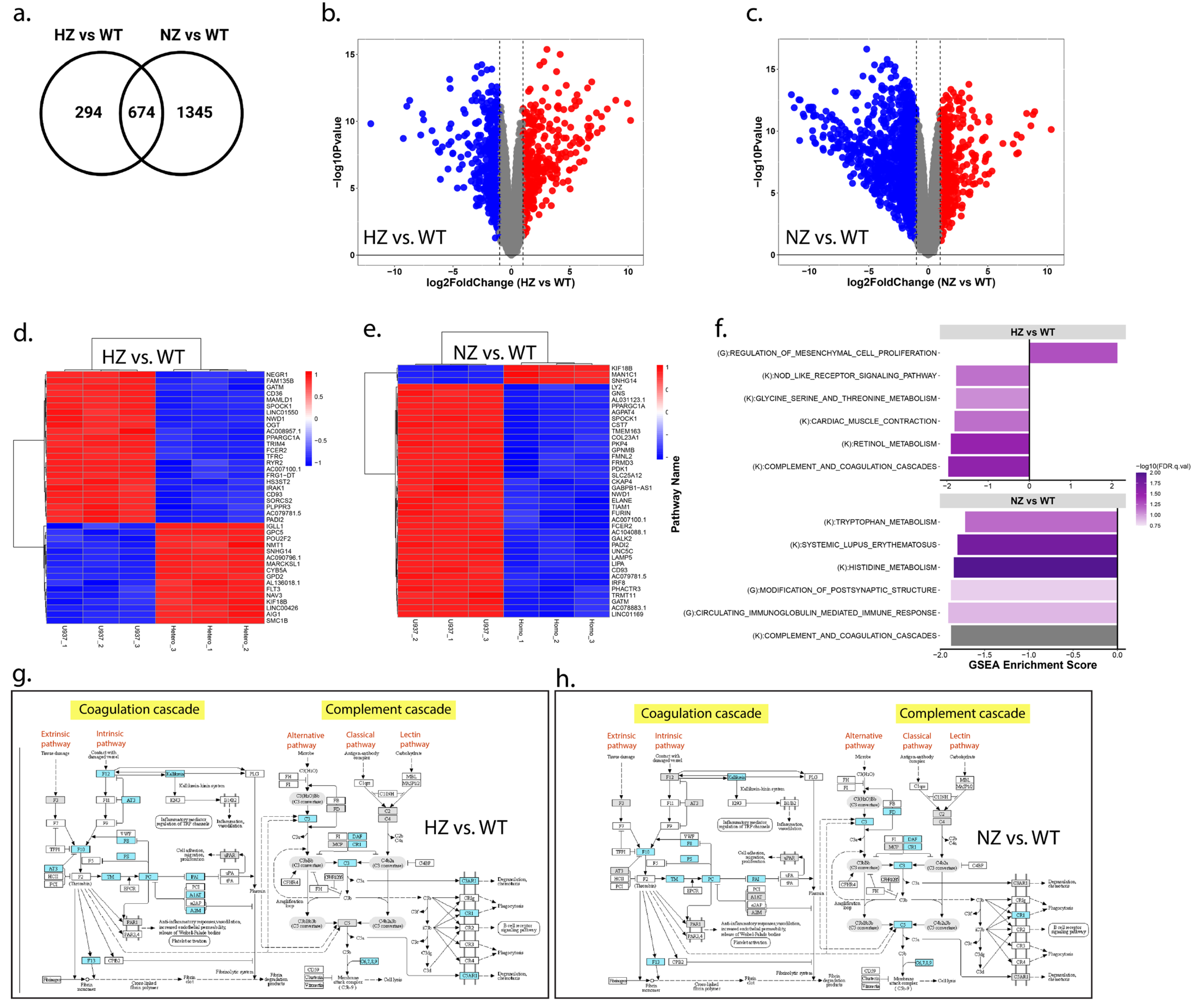
3.9.1. Loss of FURIN Impacts Transcriptomic Landscape
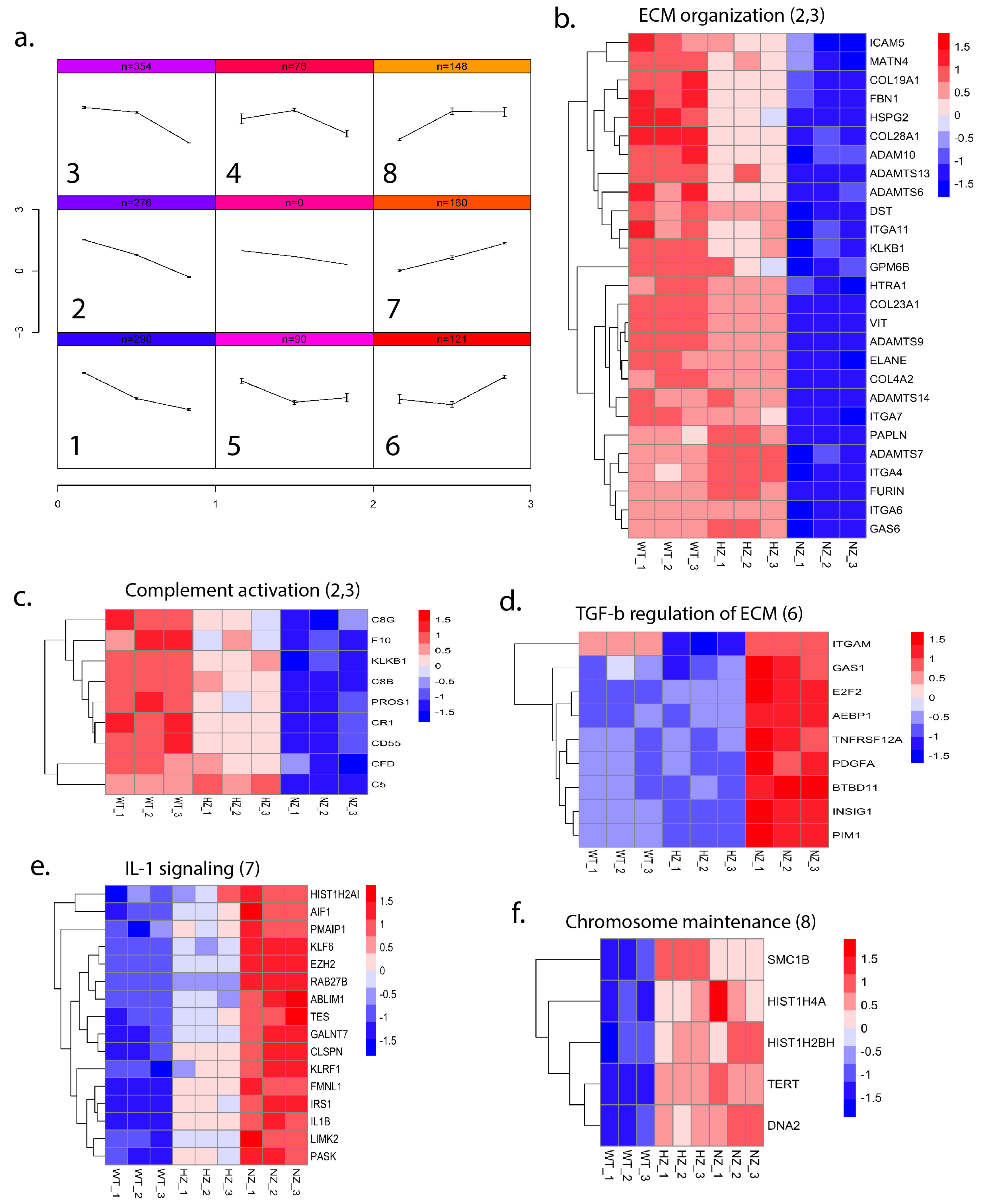
3.9.2. Loss of FURIN Affects Biological Pathways
3.9.3. Loss of FURIN Leads to Varying Gene Expression Patterns
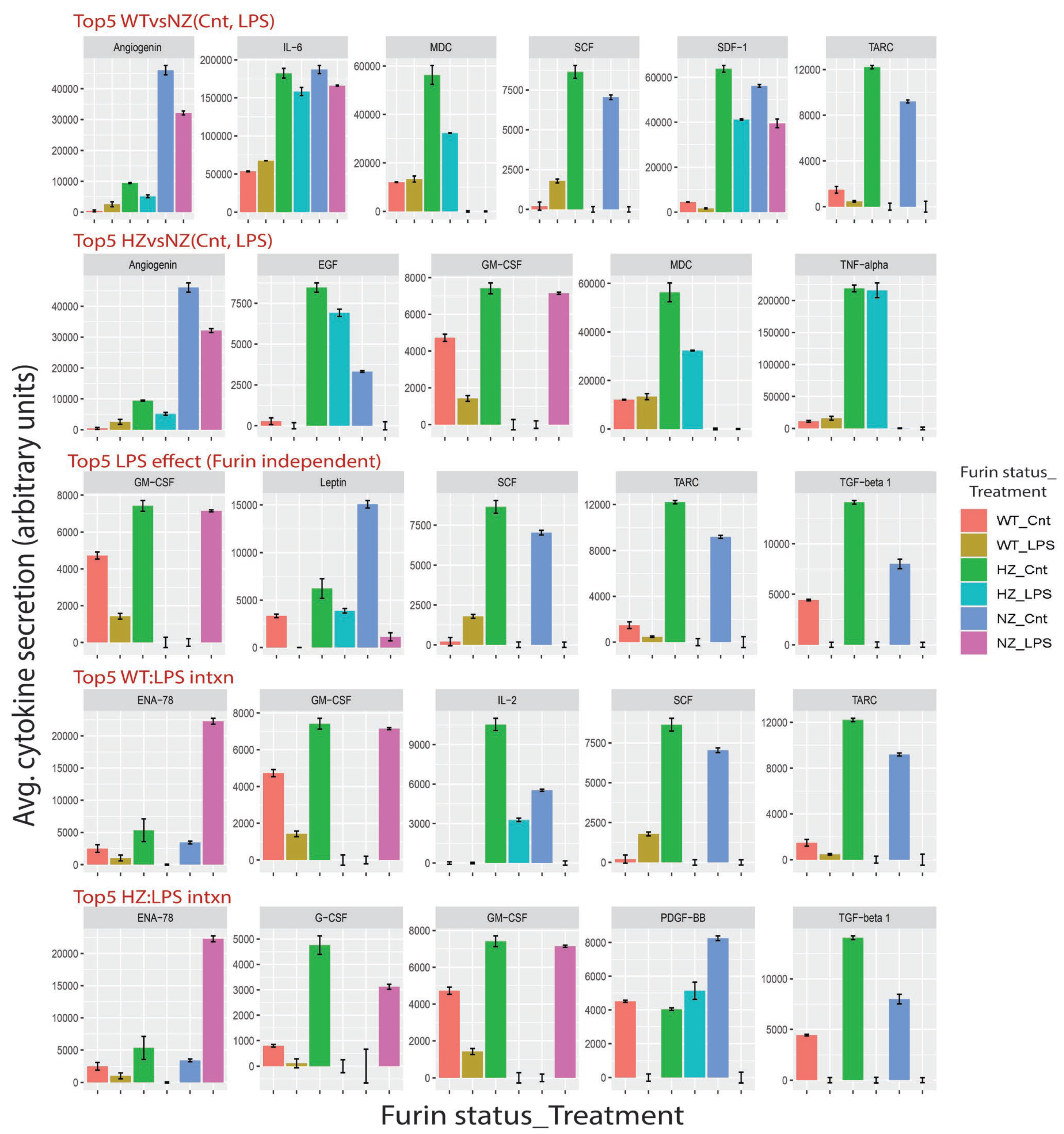
3.10. Loss of FURIN Variably Modulates Cytokine Secretion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Couture, F.; Kwiatkowska, A.; Dory, Y.L.; Day, R. Therapeutic uses of furin and its inhibitors: A patent review. Expert Opin. Ther. Pat. 2015, 25, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Jiang, T.; Zheng, X.L.; Zhao, G.J. Proprotein convertase furin/PCSK3 and atherosclerosis: New insights and potential therapeutic targets. Atherosclerosis 2017, 262, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Vivar, J.; Nelson, C.P.; Willenborg, C.; Segrè, A.V.; Mäkinen, V.-P.; Nikpay, M.; Erdmann, J.; Blankenberg, S.; O’donnell, C.; et al. Systems Genetics Analysis of Genome-Wide Association Study Reveals Novel Associations Between Key Biological Processes and Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1712–1722. [Google Scholar] [CrossRef]
- Yakala, G.K.; Cabrera-Fuentes, H.A.; Crespo-Avilan, G.E.; Rattanasopa, C.; Burlacu, A.; George, B.L.; Anand, K.; Mayan, D.C.; Corlianò, M.; Hernández-Reséndiz, S.; et al. FURIN Inhibition Reduces Vascular Remodeling and Atherosclerotic Lesion Progression in Mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Stawowy, P.; Kallisch, H.; Stawowy, N.B.P.; Stibenz, D.; Veinot, J.P.; Gräfe, M.; Seidah, N.G.; Chrétien, M.; Fleck, E.; Graf, K. Immunohistochemical localization of subtilisin/kexin-like proprotein convertases in human atherosclerosis. Virchows Arch. 2005, 446, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Do, P.T.; Nguyen, C.X.; Bui, H.T.; Tran, L.T.N.; Stacey, G.; Gillman, J.D.; Zhang, Z.J.; Stacey, M.G. Demonstration of highly efficient dual gRNA CRISPR/Cas9 editing of the homeologous GmFAD2-1A and GmFAD2-1B genes to yield a high oleic, low linoleic and alpha-linolenic acid phenotype in soybean. BMC Plant Biol. 2019, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Pathak, B.; Zhao, S.; Manoharan, M.; Srivastava, V. Dual-targeting by CRISPR/Cas9 leads to efficient point mutagenesis but only rare targeted deletions in the rice genome. 3 Biotech 2019, 9, 158. [Google Scholar] [CrossRef]
- Adikusuma, F.; Pfitzner, C.; Thomas, P.Q. Versatile single-step-assembly CRISPR/Cas9 vectors for dual gRNA expression. PLoS ONE 2017, 12, e0187236. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Hou, X.; Zuo, F.; Li, X.; Pang, Y.; Jiang, G. Application of nanoparticle-based siRNA and CRISPR/Cas9 delivery systems in gene-targeted therapy. Nanomedicine 2019, 14, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Chanput, W.; Peters, V.; Wichers, H. THP-1 and U937 Cells. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 147–159. [Google Scholar]
- Zhang, X.; Edwards, J.P.; Mosser, D.M. The expression of exogenous genes in macrophages: Obstacles and opportunities. Methods Mol. Biol. 2009, 531, 123–143. [Google Scholar] [CrossRef]
- Burke, B.; Sumner, S.; Maitland, N.; Lewis, C.E. Macrophages in gene therapy: Cellular delivery vehicles and in vivo targets. J. Leukoc. Biol. 2002, 72, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Ooi, W.F.; Qamra, A.; Cheung, A.; Ma, D.; Sundaram, G.M.; Xu, C.; Xing, M.; Poon, L.; Wang, J.; et al. HoxC5 and miR-615-3p target newly evolved genomic regions to repress hTERT and inhibit tumorigenesis. Nat. Commun. 2018, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Reiner, A.; Yekutieli, D.; Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.-T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Molloy, S.S.; Anderson, E.D.; Jean, F.; Thomas, G. Bi-cycling the furin pathway: From TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 1999, 9, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Huang, Q.; Fang, Y.; Wu, J. FurinDB: A database of 20-residue furin cleavage site motifs, substrates and their associated drugs. Int. J. Mol. Sci. 2011, 12, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zheng, M.; Yang, Y.; Gu, X.; Yang, K.; Li, M.; Liu, Y.; Zhang, Q.; Zhang, P.; Wang, Y.; et al. Furin: A Potential Therapeutic Target for COVID-19. iScience 2020, 23, 101642. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.E.J.; Reihill, J.A.; Montgomery, B.M.; Martin, S.L. Furin as a therapeutic target in cystic fibrosis airways disease. Eur. Respir. Rev. 2023, 32, 220256. [Google Scholar] [CrossRef] [PubMed]
- Preuss, M.; König, I.R.; Thompson, J.R.; Erdmann, J.; Absher, D.; Assimes, T.L.; Blankenberg, S.; Boerwinkle, E.; Chen, L.; Cupples, L.A.; et al. Design of the Coronary ARtery DIsease Genome-Wide Replication and Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ. Cardiovasc. Genet. 2010, 3, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Yang, W.; Wu, J.; Chen, B.; Yang, X.; Chen, J.; McVey, D.G.; Andreadi, C.; Gong, P.; Webb, T.R.; et al. Influence of a Coronary Artery Disease-Associated Genetic Variant on FURIN Expression and Effect of Furin on Macrophage Behavior. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Kappert, K.; Meyborg, H.; Fritzsche, J.; Urban, D.; Krüger, J.; Wellnhofer, E.; Kintscher, U.; Fleck, E.; Stawowy, P. Proprotein convertase subtilisin/kexin type 3 promotes adipose tissue-driven macrophage chemotaxis and is increased in obesity. PLoS ONE 2013, 8, e70542. [Google Scholar] [CrossRef]
- Stawowy, P.; Fleck, E. Proprotein convertases furin and PC5: Targeting atherosclerosis and restenosis at multiple levels. J. Mol. Med. 2005, 83, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, G.; Sakurada, M.; Kitagawa, K.; Kondo, T.; Takahashi, M.; Ueno, Y. Effect of FURIN SNP rs17514846 on coronary atherosclerosis in human cardiac specimens: An autopsy study of 106 cases. Leg. Med. 2022, 55, 102006. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, L.; Mi, S.; Wang, H.; Wang, C.; Jia, W.; Gong, L.; Dong, H.; Xu, B.; Jing, Y.; et al. FURIN suppresses the progression of atherosclerosis by promoting macrophage autophagy. FASEB J. 2023, 37, e22933. [Google Scholar] [CrossRef]
- Cordova, Z.M.; Grönholm, A.; Kytölä, V.; Taverniti, V.; Hämäläinen, S.; Aittomäki, S.; Niininen, W.; Junttila, I.; Ylipää, A.; Nykter, M.; et al. Myeloid cell expressed proprotein convertase FURIN attenuates inflammation. Oncotarget 2016, 7, 54392–54404. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Gruber, E.J.; Aygun, A.Y.; Leifer, C.A. Macrophage uptake of oxidized and acetylated low-density lipoproteins and generation of reactive oxygen species are regulated by linear stiffness of the growth surface. PLoS ONE 2021, 16, e0260756. [Google Scholar] [CrossRef]
- Lin, P.; Ji, H.H.; Li, Y.J.; Guo, S.D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 679797. [Google Scholar] [CrossRef]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction between the coagulation and complement system. Adv. Exp. Med. Biol. 2008, 632, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Snarski, P.; Sukhanov, S.; Yoshida, T.; Higashi, Y.; Danchuk, S.; Chandrasekar, B.; Tian, D.; Rivera-Lopez, V.; Delafontaine, P. Macrophage-Specific IGF-1 Overexpression Reduces CXCL12 Chemokine Levels and Suppresses Atherosclerotic Burden in Apoe-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sukhanov, S.; Shai, S.-Y.; Danchuk, S.; Tang, R.; Snarski, P.; Li, Z.; Lobelle-Rich, P.; Wang, M.; Wang, D.; et al. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E-Deficient Mice. Circulation 2016, 133, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Su, E.; Yu, P.; Zhang, B.; Zhang, A.; Xie, S.; Zhang, C.; Li, S.; Zou, Y.; Liu, M.; Jiang, H.; et al. Endothelial Intracellular ANG (Angiogenin) Protects Against Atherosclerosis by Decreasing Endoplasmic Reticulum Stress. Arter. Thromb. Vasc. Biol. 2022, 42, 305–325. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chua, R.; Wang, L.; Singaraja, R.; Ghosh, S. Functional and Multi-Omics Effects of an Optimized CRISPR-Mediated FURIN Depletion in U937 Monocytes. Cells 2024, 13, 588. https://doi.org/10.3390/cells13070588
Chua R, Wang L, Singaraja R, Ghosh S. Functional and Multi-Omics Effects of an Optimized CRISPR-Mediated FURIN Depletion in U937 Monocytes. Cells. 2024; 13(7):588. https://doi.org/10.3390/cells13070588
Chicago/Turabian StyleChua, Ruiming, Lijin Wang, Roshni Singaraja, and Sujoy Ghosh. 2024. "Functional and Multi-Omics Effects of an Optimized CRISPR-Mediated FURIN Depletion in U937 Monocytes" Cells 13, no. 7: 588. https://doi.org/10.3390/cells13070588