
An OX-Tra’Ordinary Tale: The Role of OX40 and OX40L in Atopic Dermatitis
 ,
,
Abstract
:1. Introduction
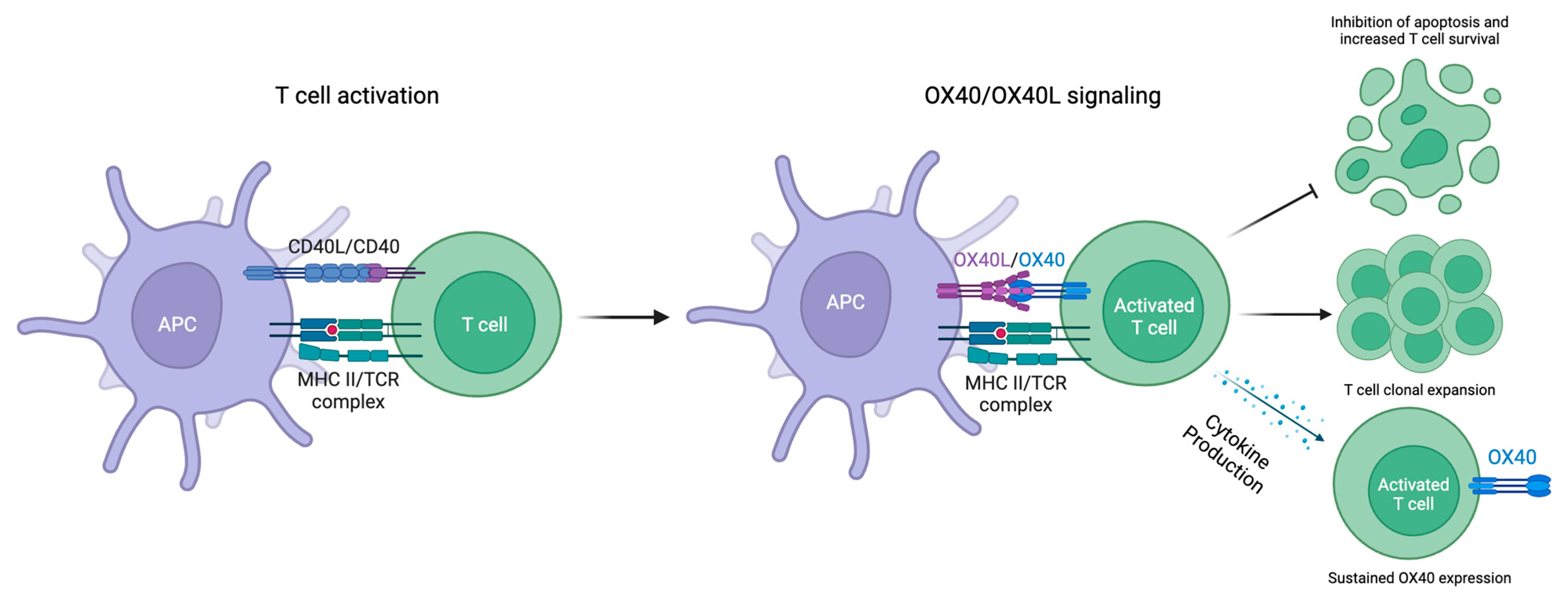
2. Overview of OX40 Signaling Pathways
2.1. OX40 Expression
2.2. OX40L Expression
2.3. OX40-OX40L Signaling
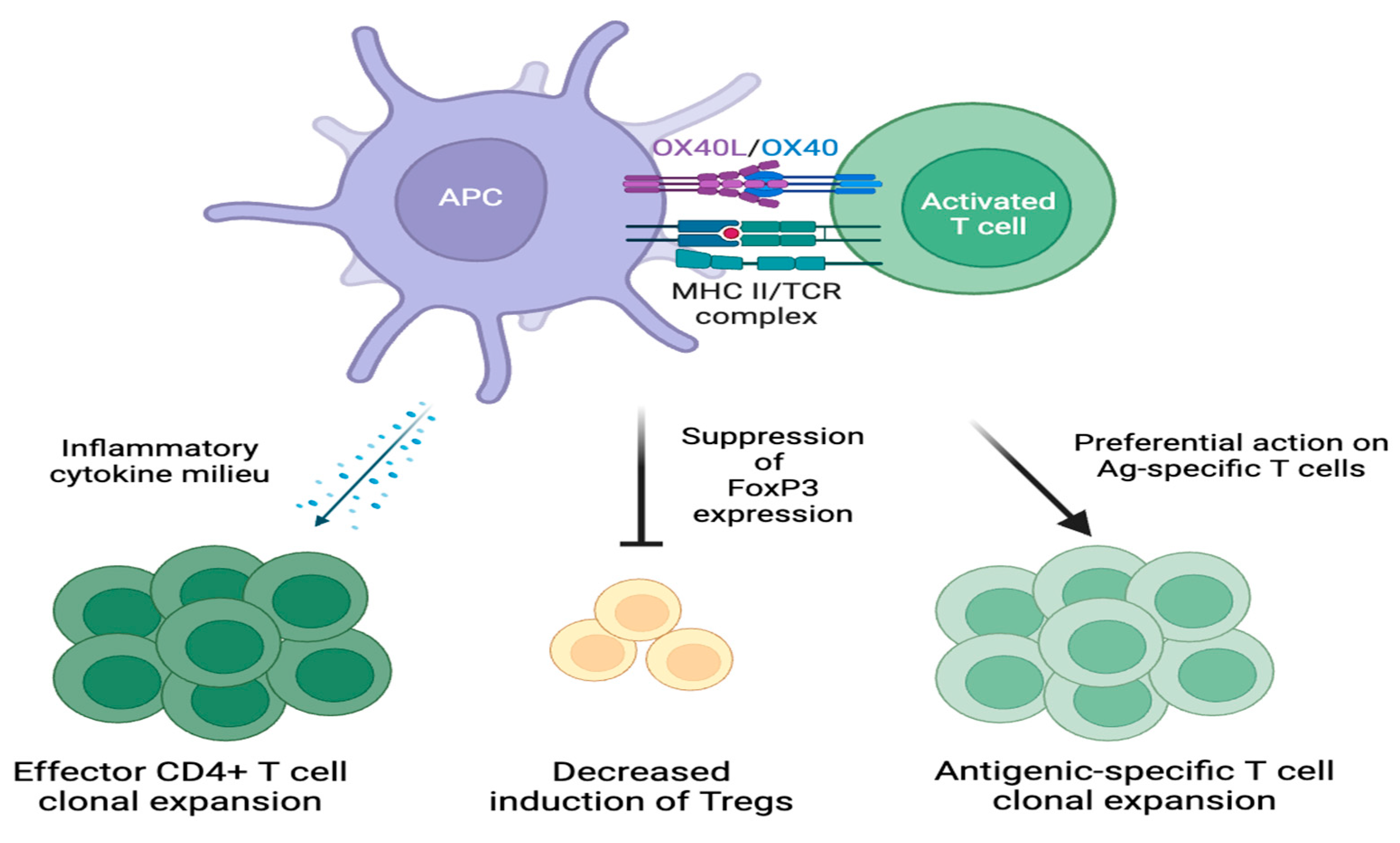
2.4. Downstream Effects of OX40-OX40L Signaling
3. AD Pathogenesis
3.1. Phenotypic Variance
3.2. Skin Barrier Dysfunction
3.3. Skin Dysbiosis
3.4. Immune System Dysregulation
4. OX40-OX40L Signaling in Atopic Dermatitis
4.1. T Helper 2 Cells
4.2. T Helper 1, T Helper 17, and T Helper 22 Cells
4.3. Memory T Cells
4.4. Suppression of Apoptosis
4.5. Modulation of OX40-OX40L Signaling
5. Therapeutic Targets and Emerging Therapies
5.1. Rocatinlimab
5.2. Telazorlimab
5.3. Amlitelimab
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Nutten, S. Atopic dermatitis: Global epidemiology and risk factors. Ann. Nutr. Metab. 2015, 66, 8–16. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Krueger, J.G.; Lebwohl, M.G. Systemic immune mechanisms in atopic dermatitis and psoriasis with implications for treatment. Exp. Dermatol. 2018, 27, 409–417. [Google Scholar] [CrossRef]
- Ständer, S. Atopic Dermatitis. N. Engl. J. Med. 2021, 384, 1136–1143. [Google Scholar] [CrossRef]
- Itamura, M.; Sawada, Y. Involvement of Atopic Dermatitis in the Development of Systemic Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 13445. [Google Scholar] [CrossRef]
- Furue, M.; Kadono, T. “Inflammatory skin march” in atopic dermatitis and psoriasis. Inflamm. Res. 2017, 66, 833–842. [Google Scholar] [CrossRef]
- Cesare, A.D.; Meglio, P.D.; Nestle, F.O. A role for Th17 cells in the immunopathogenesis of atopic dermatitis? J. Investig. Dermatol. 2008, 128, 2569–2571. [Google Scholar] [CrossRef]
- Bieber, T. Atopic dermatitis: An expanding therapeutic pipeline for a complex disease. Nat. Rev. Drug. Discov. 2022, 21, 21–40. [Google Scholar] [CrossRef]
- Facheris, P.; Jeffery, J.; Del Duca, E.; Guttman-Yassky, E. The translational revolution in atopic dermatitis: The paradigm shift from pathogenesis to treatment. Cell. Mol. Immunol. 2023, 20, 448–474. [Google Scholar] [CrossRef]
- Mikhaylov, D.; Ungar, B.; Renert-Yuval, Y.; Guttman-Yassky, E. Oral Janus kinase inhibitors for atopic dermatitis. Ann. Allergy Asthma. Immunol. 2023, 130, 577–592. [Google Scholar] [CrossRef]
- Croft, M.; Esfandiari, E.; Chong, C.; Hsu, H.; Kabashima, K.; Kricorian, G.; Warren, R.B.; Wollenberg, A.; Guttman-Yassky, E. OX40 in the Pathogenesis of Atopic Dermatitis—A New Therapeutic Target. Am. J. Clin. Dermatol. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.J.; Hirschfield, G.M.; Lane, P.J. OX40, OX40L and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Duttagupta, P.A.; Boesteanu, A.C.; Katsikis, P.D. Costimulation signals for memory CD8+ T cells during viral infections. Crit. Rev. Immunol. 2009, 29, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Furue, M. OX40L-OX40 Signaling in Atopic Dermatitis. J. Clin. Med. 2021, 10, 2578. [Google Scholar] [CrossRef] [PubMed]
- Alves Costa Silva, C.; Facchinetti, F.; Routy, B.; Derosa, L. New pathways in immune stimulation: Targeting OX40. ESMO Open 2020, 5, e000573. [Google Scholar] [CrossRef] [PubMed]
- Ward-Kavanagh, L.K.; Lin, W.W.; Šedý, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhao, S.; Zhang, X.; Jia, K.; Wang, H.; Zhou, C.; He, Y. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. Onco. Targets. Ther. 2019, 12, 7347–7353. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Salek-Ardakani, S.; Song, J.; So, T.; Bansal-Pakala, P. Regulation of T Cell Immunity by OX40 and OX40L. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2000–2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK5990/ (accessed on 25 January 2024).
- Redmond, W.L.; Ruby, C.E.; Weinberg, A.D. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit. Rev. Immunol. 2009, 29, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Iriki, H.; Takahashi, H.; Amagai, M. Diverse Role of OX40 on T Cells as a Therapeutic Target for Skin Diseases. J. Investig. Dermatol. 2023, 143, 545–553. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Simpson, E.L.; Reich, K.; Kabashima, K.; Igawa, K.; Suzuki, T.; Mano, H.; Matsui, T.; Esfandiari, E.; Furue, M. An anti-OX40 antibody to treat moderate-to-severe atopic dermatitis: A multicentre, double-blind, placebo-controlled phase 2b study. Lancet 2023, 401, 204–214. [Google Scholar] [CrossRef]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef]
- Stüber, E.; Neurath, M.; Calderhead, D.; Fell, H.P.; Strober, W. Cross-linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity 1995, 2, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, Y.; Tanaka, Y.; Tozawa, H.; Takahashi, Y.; Maliszewski, C.; Delespesse, G. Expression and function of OX40 ligand on human dendritic cells. J. Immunol. 1997, 159, 3838–3848. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Amakawa, R.; Inaba, M.; Hori, T.; Ota, M.; Nakamura, K.; Takebayashi, M.; Miyaji, M.; Yoshimura, T.; Inaba, K.; et al. Plasmacytoid dendritic cells regulate Th cell responses through OX40 ligand and type I IFNs. J. Immunol. 2004, 172, 4253–4259. [Google Scholar] [CrossRef]
- Sato, T.; Ishii, N.; Murata, K.; Kikuchi, K.; Nakagawa, S.; Ndhlovu, L.C.; Sugamura, K. Consequences of OX40-OX40 ligand interactions in langerhans cell function: Enhanced contact hypersensitivity responses in OX40L-transgenic mice. Eur. J. Immunol. 2002, 32, 3326–3335. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Wegmann, K.W.; Funatake, C.; Whitham, R.H. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J. Immunol. 1999, 162, 1818–1826. [Google Scholar] [CrossRef]
- Nakagawa, H.; Iizuka, H.; Nemoto, O.; Shimabe, M.; Furukawa, Y.; Kikuta, N.; Ootaki, K. Safety, tolerability and efficacy of repeated intravenous infusions of KHK4083, a fully human anti-OX40 monoclonal antibody, in Japanese patients with moderate to severe atopic dermatitis. J. Dermatol. Sci. 2020, 99, 82–89. [Google Scholar] [CrossRef]
- Zheng, C.; Shi, Y.; Zou, Y. T cell co-stimulatory and co-inhibitory pathways in atopic dermatitis. Front. Immunol. 2023, 14, 1081999. [Google Scholar] [CrossRef] [PubMed]
- Imura, A.; Hori, T.; Imada, K.; Ishikawa, T.; Tanaka, Y.; Maeda, M.; Imamura, S.; Uchiyama, T. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J. Exp. Med. 1996, 183, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.K.; Carlin, S.; Pack, R.A.; Arndt, G.M.; Au, W.W.; Johnson, P.R.; Black, J.L.; Hunt, N.H. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: A possible role in asthma? J. Allergy Clin. Immunol. 2004, 113, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Kashiwakura, J.; Yokoi, H.; Saito, H.; Okayama, Y. T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: Comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J. Immunol. 2004, 173, 5247–5257. [Google Scholar] [CrossRef] [PubMed]
- Gough, M.J.; Weinberg, A.D. OX40 (CD134) and OX40L. Adv. Exp. Med. Biol. 2009, 647, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.O.; Rittenhouse, S.E.; Tsichlis, P.N. AKT/PKB and other D3 phosphoinositide-regulated kinases: Kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 1999, 68, 965–1014. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; So, T.; Croft, M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J. Immunol. 2008, 180, 7240–7248. [Google Scholar] [CrossRef]
- Kane, L.P.; Weiss, A. The PI-3 kinase/Akt pathway and T cell activation: Pleiotropic pathways downstream of PIP3. Immunol. Rev. 2003, 192, 7–20. [Google Scholar] [CrossRef]
- Rudensky, A. Foxp3 and dominant tolerance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1645–1646. [Google Scholar] [CrossRef]
- Vu, M.D.; Xiao, X.; Gao, W.; Degauque, N.; Chen, M.; Kroemer, A.; Killeen, N.; Ishii, N.; Li, X.C. OX40 costimulation turns off Foxp3+ Tregs. Blood 2007, 110, 2501–2510. [Google Scholar] [CrossRef]
- Gramaglia, I.; Jember, A.; Pippig, S.D.; Weinberg, A.D.; Killeen, N.; Croft, M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000, 165, 3043–3050. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Ishii, N.; Murata, K.; Sato, T.; Sugamura, K. Critical involvement of OX40 ligand signals in the T cell priming events during experimental autoimmune encephalomyelitis. J. Immunol. 2001, 167, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Ruby, C.E.; Yates, M.A.; Hirschhorn-Cymerman, D.; Chlebeck, P.; Wolchok, J.D.; Houghton, A.N.; Offner, H.; Weinberg, A.D. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 2009, 183, 4853–4857. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas Krohn, I.; Aerts, J.L.; Breckpot, K.; Goyvaerts, C.; Knol, E.; Van Wijk, F.; Gutermuth, J. T-cell subsets in the skin and their role in inflammatory skin disorders. Allergy 2022, 77, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I. Atopic Dermatitis in Adults. Med. Clin. N. Am. 2020, 104, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Guttman-Yassky, E. Deciphering the complexities of atopic dermatitis: Shifting paradigms in treatment approaches. J. Allergy Clin. Immunol. 2014, 134, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Koh, L.F.; Ong, R.Y.; Common, J.E. Skin microbiome of atopic dermatitis. Allergol. Int. 2022, 71, 31–39. [Google Scholar] [CrossRef]
- Galli, E.; Cinicola, B.; Carello, R.; Caimmi, S.; Brindisi, G.; De Castro, G.; Zicari, A.M.; Tosca, M.A.; Manti, S.; Martelli, A.; et al. Atopic dermatitis. Acta Biomed. 2020, 91, e2020011. [Google Scholar] [CrossRef]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Bakker, D.; de Bruin-Weller, M.; Drylewicz, J.; van Wijk, F.; Thijs, J. Biomarkers in atopic dermatitis. J. Allergy Clin. Immunol. 2023, 151, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Leonard, A.; Pavel, A.B.; Malik, K.; Raja, A.; Glickman, J.; Estrada, Y.D.; Peng, X.; Del Duca, E.; Sanz-Cabanillas, J.; et al. Age-specific changes in the molecular phenotype of patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Wakefield, J.S. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 781–791.e1. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y. Clinical implications of new mechanistic insights into atopic dermatitis. Curr. Opin. Pediatr. 2016, 28, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, B.E.; Leung, D.Y.M. Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma. Proc. 2019, 40, 84–92. [Google Scholar] [CrossRef]
- Fyhrquist, N.; Muirhead, G.; Prast-Nielsen, S.; Jeanmougin, M.; Olah, P.; Skoog, T.; Jules-Clement, G.; Feld, M.; Barrientos-Somarribas, M.; Sinkko, H.; et al. Microbe-host interplay in atopic dermatitis and psoriasis. Nat. Commun. 2019, 10, 4703. [Google Scholar] [CrossRef] [PubMed]
- Hrestak, D.; Matijašić, M.; Čipčić Paljetak, H.; Ledić Drvar, D.; Ljubojević Hadžavdić, S.; Perić, M. Skin Microbiota in Atopic Dermatitis. Int. J. Mol. Sci. 2022, 23, 3503. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, R.D.; Bandier, J.; Skov, L.; Engstrand, L.; Johansen, J.D. The role of the skin microbiome in atopic dermatitis: A systematic review. Br. J. Dermatol. 2017, 177, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, D.; Grywalska, E. The Role of Immune Defects and Colonization of Staphylococcus aureus in the Pathogenesis of Atopic Dermatitis. Anal. Cell. Pathol. 2018, 2018, 1956403. [Google Scholar] [CrossRef]
- Williams, M.R.; Nakatsuji, T.; Sanford, J.A.; Vrbanac, A.F.; Gallo, R.L. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J. Investig. Dermatol. 2017, 137, 377–384. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Investig. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Colafrancesco, S.; Emmi, G.; Imazio, M.; Lopalco, G.; Maggio, M.C.; Sota, J.; Dinarello, C.A. Interleukin 1α: A comprehensive review on the role of IL-1α in the pathogenesis and treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2021, 20, 102763. [Google Scholar] [CrossRef] [PubMed]
- Mesjasz, A.; Trzeciak, M.; Gleń, J.; Jaskulak, M. Potential Role of IL-37 in Atopic Dermatitis. Cells 2023, 12, 2766. [Google Scholar] [CrossRef] [PubMed]
- Kunimura, K.; Fukui, Y. The molecular basis for IL-31 production and IL-31-mediated itch transmission: From biology to drug development. Int. Immunol. 2021, 33, 731–736. [Google Scholar] [CrossRef]
- Fania, L.; Moretta, G.; Antonelli, F.; Scala, E.; Abeni, D.; Albanesi, C.; Madonna, S. Multiple Roles for Cytokines in Atopic Dermatitis: From Pathogenic Mediators to Endotype-Specific Biomarkers to Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 2684. [Google Scholar] [CrossRef] [PubMed]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell-derived cytokines: More than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef]
- Furue, M.; Ulzii, D.; Vu, Y.H.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of Atopic Dermatitis: Current Paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar] [CrossRef]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Ellis, C.N.; Mancini, A.J.; Paller, A.S.; Simpson, E.L. Atopic dermatitis: Epidemiology and pathogenesis update. Semin. Cutan. Med. Surg. 2012, 31, S3–S5. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Bansal-Pakala, P.; Jember, A.G.; Croft, M. Signaling through OX40 (CD134) breaks peripheral T-cell tolerance. Nat. Med. 2001, 7, 907–912. [Google Scholar] [CrossRef]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Waldman, A.; Ahluwalia, J.; Ong, P.Y.; Eichenfield, L.F. Atopic dermatitis: Pathogenesis. Semin. Cutan. Med. Surg. 2017, 36, 100–103. [Google Scholar] [CrossRef]
- Fu, N.; Xie, F.; Sun, Z.; Wang, Q. The OX40/OX40L Axis Regulates T Follicular Helper Cell Differentiation: Implications for Autoimmune Diseases. Front. Immunol. 2021, 12, 670637. [Google Scholar] [CrossRef]
- Hudak, S.; Hagen, M.; Liu, Y.; Catron, D.; Oldham, E.; McEvoy, L.M.; Bowman, E.P. Immune surveillance and effector functions of CCR10(+) skin homing T cells. J. Immunol. 2002, 169, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shen, Z. Tissue-resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell. Mol. Immunol. 2020, 17, 64–75. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold. Spring. Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold. Spring. Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell. Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Akdis, M.; Trautmann, A.; Blaser, K. Immune regulation in atopic dermatitis. Curr. Opin. Immunol. 2000, 12, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Ferran, M.; Santamaria-Babi, L.F. Pathological mechanisms of skin homing T cells in atopic dermatitis. World Allergy Organ. J. 2010, 3, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; So, T.; Cheng, M.; Tang, X.; Croft, M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity 2005, 22, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Gooderham, M.J.; Girard, G.; Raman, M.; Strout, V. Phase I randomized study of KHK4083, an anti-OX40 monoclonal antibody, in patients with mild to moderate plaque psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1324–1332. [Google Scholar] [CrossRef]
- Lé, A.M.; Torres, T. OX40-OX40L Inhibition for the Treatment of Atopic Dermatitis-Focus on Rocatinlimab and Amlitelimab. Pharmaceutics 2022, 14, 2753. [Google Scholar] [CrossRef]
- Rewerska, B.; Sher, L.D.; Alpizar, S.; Pauser, S.; Pulka, G.; Mozaffarian, N.; Salhi, Y.; Martinet, C.; Jabert, W.; Gudi, G.; et al. Phase 2b randomized trial of OX40 inhibitor telazorlimab for moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. Glob. 2023, 3, 100195. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Pavel, A.B.; Zhou, L.; Estrada, Y.D.; Zhang, N.; Xu, H.; Peng, X.; Wen, H.C.; Govas, P.; Gudi, G.; et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 482–493.e7. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Bieber, T.; Cork, M.J.; Reich, A.; Wilson, R.; Quaratino, S.; Stebegg, M.; Brennan, N.; Gilbert, S.; O’Malley, J.T.; et al. Safety and efficacy of amlitelimab, a fully human nondepleting, noncytotoxic anti-OX40 ligand monoclonal antibody, in atopic dermatitis: Results of a phase IIa randomized placebo-controlled trial. Br. J. Dermatol. 2023, 189, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Blauvelt, A.; Papp, K.; Reich, A.; Lee, C.-H.; Worm, M.; Lynde, C.; Kataoka, Y.; Foley, P.; Weber, C.; et al. 227 Efficacy and safety of amlitelimab (an anti-OX40 ligand antibody) in patients with moderate-to-severe atopic dermatitis: 24-week results from a Phase 2b trial (STREAM-AD). J. Investig. Dermatol. 2023, 143, S370. [Google Scholar] [CrossRef]
- Akinlade, B.; Guttman-Yassky, E.; de Bruin-Weller, M.; Simpson, E.L.; Blauvelt, A.; Cork, M.J.; Prens, E.; Asbell, P.; Akpek, E.; Corren, J.; et al. Conjunctivitis in dupilumab clinical trials. Br. J. Dermatol. 2019, 181, 459–473. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Toth, D.; Bieber, T.; Alexis, A.F.; Elewski, B.E.; Pink, A.E.; Hijnen, D.; Jensen, T.N.; Bang, B.; Olsen, C.K.; et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: Results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br. J. Dermatol. 2021, 184, 450–463. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent | Site of Action | Study Design | Primary Endpoint | Study Arms | Results | |
|---|---|---|---|---|---|---|
| Rocatinlimab | OX40 | NCT03703102 [23] Phase 2b multi-center, double-blind, placebo-controlled trial of 274 adults with moderate-to-severe AD randomized 1:1:1:1:1 | Least-squares mean percent change in EASI from baseline to week 16 | Placebo | −15.0% (95%CI, −28.6%–−1.4%) | |
| No LD 150 mg q4weeks | −48.3% (95%CI, −62.2%–−34.0%) p = 0.0003 | |||||
| No LD 600 mg q4weeks | −49.7% (95%CI, −64.3%–−35.2%) p = 0.0002 | |||||
| No LD 300 mg q2weeks | −61.1% (95%CI, −75.2%–−47.0%) p < 0.0001 | |||||
| No LD 600 mg q2weeks | −57.4% (95%CI, −71.3%–−43.4%) p < 0.0001 | |||||
| Telazorlimab | OX40 | NCT03568162 [93] Phase 2b multi-center, double-blind, placebo-controlled trial of adults with moderate-to-severe AD | Part 1 313 subjects randomized 1:1:1:1 | Least-squares mean percent change in EASI from baseline to week 16 | Placebo | −34.2% (SE 5.5) |
| LD: 150 mg 75 mg q4weeks | −31.0% (SE 5.7) p = 0.691 | |||||
| LD: 600 mg 300 mg q4weeks | −48.6% (SE 5.4) p = 0.061 | |||||
| LD: 600 mg 300 mg q2weeks | −54.4% (SE 5.1) p = 0.008 | |||||
| Part 2 149 subjects randomized 1:1 | Least-squares mean percent change in EASI from baseline to week 16 | Placebo | −41.8% (SE 4.7) | |||
| LD: 1200 mg 600 mg q2weeks | −59.0% (SE 4.6) p = 0.008 | |||||
| Amlitelimab | OX40L | NCT05131477 [95] Phase 2b multi-center, double-blind, placebo-controlled trial of 390 adults with moderate-to-severe AD randomized 1:1:1:1:1 | Least-squares mean percent change in EASI from baseline to week 16 | Placebo | −29.4% | |
| LD: 500 mg 250 mg q4weeks | −61.5% p < 0.0001 | |||||
| No LD 250 mg q4weeks | −56.8% p < 0.0001 | |||||
| No LD 125 mg q4weeks | −51.6% p = 0.0002 | |||||
| No LD 62.5 mg q4weeks | −59.6% p < 0.0001 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadrolashrafi, K.; Guo, L.; Kikuchi, R.; Hao, A.; Yamamoto, R.K.; Tolson, H.C.; Bilimoria, S.N.; Yee, D.K.; Armstrong, A.W. An OX-Tra’Ordinary Tale: The Role of OX40 and OX40L in Atopic Dermatitis. Cells 2024, 13, 587. https://doi.org/10.3390/cells13070587
Sadrolashrafi K, Guo L, Kikuchi R, Hao A, Yamamoto RK, Tolson HC, Bilimoria SN, Yee DK, Armstrong AW. An OX-Tra’Ordinary Tale: The Role of OX40 and OX40L in Atopic Dermatitis. Cells. 2024; 13(7):587. https://doi.org/10.3390/cells13070587
Chicago/Turabian StyleSadrolashrafi, Kaviyon, Lily Guo, Robin Kikuchi, Audrey Hao, Rebecca K. Yamamoto, Hannah C. Tolson, Sara N. Bilimoria, Danielle K. Yee, and April W. Armstrong. 2024. "An OX-Tra’Ordinary Tale: The Role of OX40 and OX40L in Atopic Dermatitis" Cells 13, no. 7: 587. https://doi.org/10.3390/cells13070587