
The Molecular Mechanisms in Senescent Cells Induced by Natural Aging and Ionizing Radiation
 , , ,
, , ,
Abstract
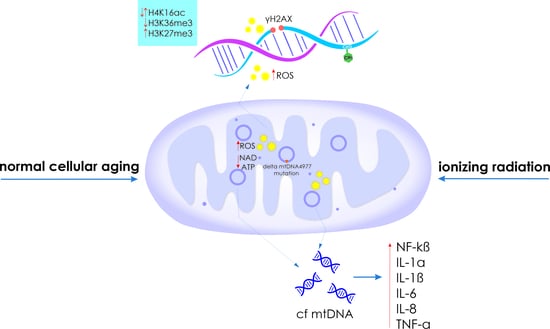
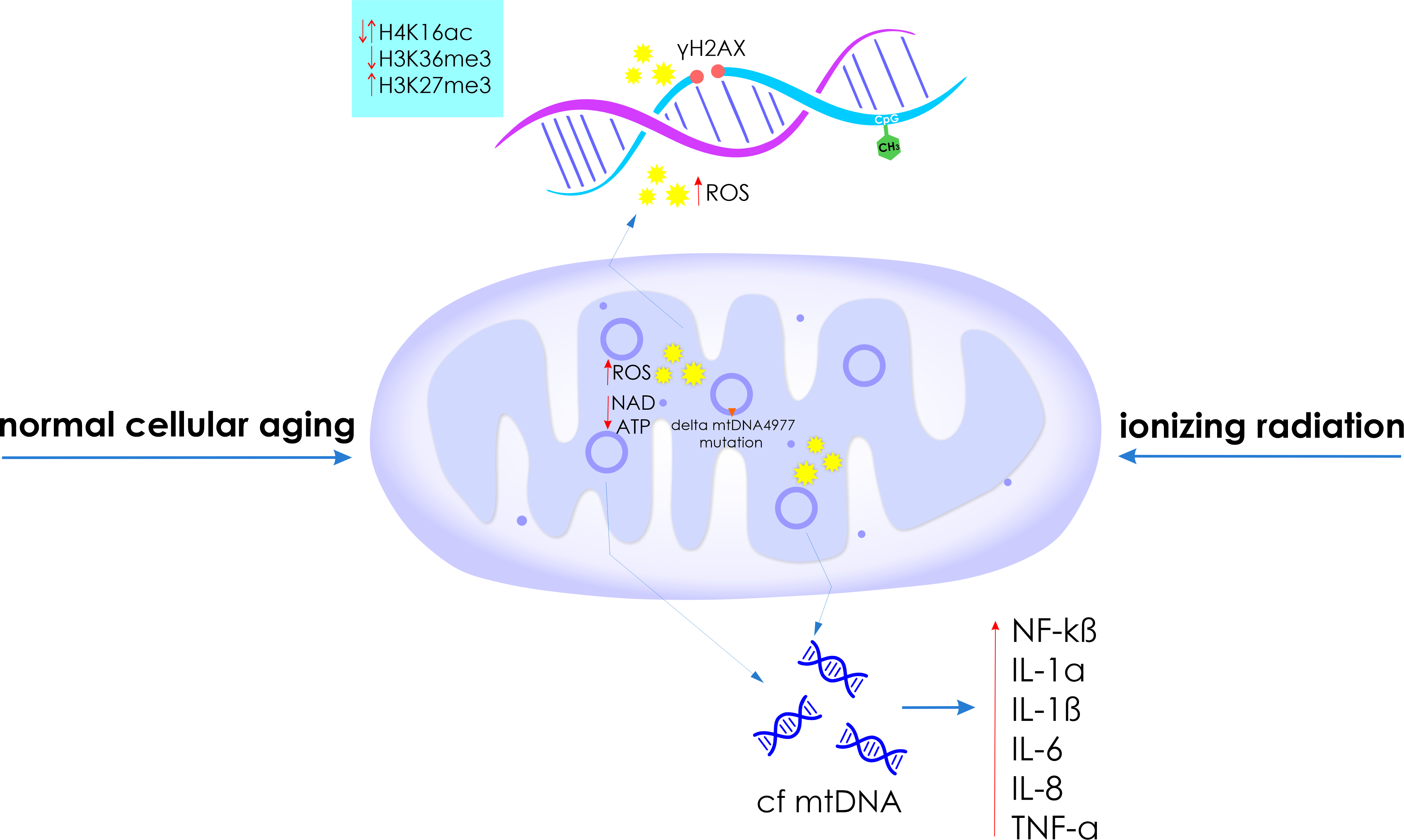
:
1. Introduction
2. Inducers and Key Signatures of Cellular Senescence
2.1. Genome Instability
2.2. Disruption of Mitochondrial Profile
2.3. miRNAs in the Mechanism of Cellular Senescence
2.4. Epigenetic Changes
3. Senescence-Associated Secretory Phenotype (SASP)
3.1. Transcription Factor NF-kB
3.2. Cytokines
4. Radiation-Induced Cellular Senescence
4.1. DNA Damage
4.2. Mitochondrial Dysfunction
4.3. Associated miRNAs
4.4. DNA Methylation and Histone Modification
5. Ionizing Radiation and SASP
5.1. Role of IR in the Induction of NF-kB
5.2. TNF-α, IL-1α, IL-1β, IL-6, and IL-8
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and Aging-Related Diseases: From Molecular Mechanisms to Interventions and Treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- Al-Jumayli, M.; Brown, S.L.; Chetty, I.J.; Extermann, M.; Movsas, B. The Biological Process of Aging and the Impact of Ionizing Radiation. Semin. Radiat. Oncol. 2022, 32, 172–178. [Google Scholar] [CrossRef]
- Zhao, L.; Bao, C.; Shang, Y.; He, X.; Ma, C.; Lei, X.; Mi, D.; Sun, Y. The Determinant of DNA Repair Pathway Choices in Ionising Radiation-Induced DNA Double-Strand Breaks. Biomed. Res. Int. 2020, 2020, 4834965. [Google Scholar] [CrossRef]
- Wang, Q.-Q.; Yin, G.; Huang, J.-R.; Xi, S.-J.; Qian, F.; Lee, R.-X.; Peng, X.-C.; Tang, F.-R. Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms. Cells 2021, 10, 3570. [Google Scholar] [CrossRef] [PubMed]
- Alessio, N.; Del Gaudio, S.; Capasso, S.; Di Bernardo, G.; Cappabianca, S.; Cipollaro, M.; Peluso, G.; Galderisi, U. Low Dose Radiation Induced Senescence of Human Mesenchymal Stromal Cells and Impaired the Autophagy Process. Oncotarget 2015, 6, 8155–8166. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Minami, M.; Yoshida, K.; Nagata, M.; Miyata, T.; Yang, T.; Takayama, N.; Suzuki, K.; Okawa, M.; Yamada, K.; et al. Irradiation Accelerates Plaque Formation and Cellular Senescence in Flow-Altered Carotid Arteries of Apolipoprotein E Knock-Out Mice. J. Am. Heart Assoc. 2021, 10, e020712. [Google Scholar] [CrossRef] [PubMed]
- Livingston, K.; Schlaak, R.A.; Puckett, L.L.; Bergom, C. The Role of Mitochondrial Dysfunction in Radiation-Induced Heart Disease: From Bench to Bedside. Front. Cardiovasc. Med. 2020, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Dondoladze, K.; Nikolaishvili, M.; Museliani, T.; Jikia, G. EFFECT OF RADIATION ON AGING PROCESSES AND TELOMERE LENGTH. Probl. Radiat. Med. Radiobiol. 2022, 27, 107–119. [Google Scholar] [CrossRef]
- Robert, G.; Wagner, J.R. ROS-Induced DNA Damage as an Underlying Cause of Aging. Adv. Geriatr. Med. Res. 2020, 2, e200024. [Google Scholar] [CrossRef]
- Zlotorynski, E. Defective Mitochondria Ignite the SASP. Nat. Rev. Mol. Cell Biol. 2020, 21, 179. [Google Scholar] [CrossRef]
- Hoyes, K.P.; Hendry, J.H.; Lord, B.I. Modification of Murine Adult Haemopoiesis and Response to Methyl Nitrosourea Following Exposure to Radiation at Different Developmental Stages. Int. J. Radiat. Biol. 2000, 76, 77–85. [Google Scholar] [CrossRef]
- Hernández, L.; Terradas, M.; Camps, J.; Martín, M.; Tusell, L.; Genescà, A. Aging and Radiation: Bad Companions. Aging Cell 2015, 14, 153–161. [Google Scholar] [CrossRef]
- Ozasa, K. Epidemiological Research on Radiation-Induced Cancer in Atomic Bomb Survivors. J. Radiat. Res. 2016, 57, i112–i117. [Google Scholar] [CrossRef]
- Mohamad Kamal, N.S.; Safuan, S.; Shamsuddin, S.; Foroozandeh, P. Aging of the Cells: Insight into Cellular Senescence and Detection Methods. Eur. J. Cell Biol. 2020, 99, 151108. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular Senescence in Ageing: From Mechanisms to Therapeutic Opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yang, X.; Liu, X. Resveratrol Attenuates Cigarette Smoke Extract Induced Cellular Senescence in Human Airway Epithelial Cells by Regulating the MiR-34a/SIRT1/NF-ΚB Pathway. Medicine 2022, 101, e31944. [Google Scholar] [CrossRef]
- Liu, C.-W.; Chen, P.-H.; Yu, T.-J.; Lin, K.-J.; Chang, L.-C. WWOX Modulates ROS-Dependent Senescence in Bladder Cancer. Molecules 2022, 27, 7388. [Google Scholar] [CrossRef]
- Zhang, W.; Mei, R.; Wu, S. Ketorolac Tromethamine Represses Senescence in Aging Articular Chondrocytes. Biosci. Biotechnol. Biochem. 2022, 86, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhou, Y.; He, S.; Vashisth, M.K.; Jia, H.; Dai, Q.; He, Y.; Wang, X. Amonafide Induces HUVEC Senescence by Inhibiting Autophagy. Discov. Med. 2023, 35, 264. [Google Scholar] [CrossRef]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Liu, H.-L.; Nan, H.; Zhao, W.-W.; Wan, X.-B.; Fan, X.-J. Phase Separation in DNA Double-Strand Break Response. Nucleus 2024, 15, 2296243. [Google Scholar] [CrossRef]
- Sławińska, N.; Krupa, R. Molecular Aspects of Senescence and Organismal Ageing—DNA Damage Response, Telomeres, Inflammation and Chromatin. Int. J. Mol. Sci. 2021, 22, 590. [Google Scholar] [CrossRef]
- Matsuda, S.; Ikura, T.; Matsuda, T. Absolute Quantification of DNA Damage Response Proteins. Genes. Environ. 2023, 45, 37. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.M.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres Are Favoured Targets of a Persistent DNA Damage Response in Ageing and Stress-Induced Senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Giunta, S.; Fenech, M.; Neri, M.; Bonassi, S. γH2AX as a Marker of DNA Double Strand Breaks and Genomic Instability in Human Population Studies. Mutat. Res./Rev. Mutat. Res. 2013, 753, 24–40. [Google Scholar] [CrossRef]
- Kulaberoglu, Y.; Gundogdu, R.; Hergovich, A. The Role of P53/P21/P16 in DNA-Damage Signaling and DNA Repair. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 243–256. [Google Scholar]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; François, M.; Fenech, M.F.; Leifert, W.R. Persistent γH2AX: A Promising Molecular Marker of DNA Damage and Aging. Mutat. Res./Rev. Mutat. Res. 2015, 766, 1–19. [Google Scholar] [CrossRef]
- Nelson, C.B.; Alturki, T.M.; Luxton, J.J.; Taylor, L.E.; Maranon, D.G.; Muraki, K.; Murnane, J.P.; Bailey, S.M. Telomeric Double Strand Breaks in G1 Human Cells Facilitate Formation of 5′ C-Rich Overhangs and Recruitment of TERRA. Front. Genet. 2021, 12, 644803. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Mathias, B.; O’Leary, D.; Saucier, N.; Ahmad, F.; White, L.S.; Russell, L.; Shinawi, M.; Smith, M.J.; Abraham, R.S.; Cooper, M.A.; et al. MYSM1 Attenuates DNA Damage Signals Triggered by Physiologic and Genotoxic DNA Breaks. J. Allergy Clin. Immunol. 2023, in press. [CrossRef] [PubMed]
- Pustovalova, M.; Grekhova, A.; Astrelina, T.; Nikitina, V.; Dobrovolskaya, E.; Suchkova, Y.; Kobzeva, I.; Usupzhanova, D.; Vorobyeva, N.; Samoylov, A.; et al. Accumulation of Spontaneous γH2AX Foci in Long-Term Cultured Mesenchymal Stromal Cells. Aging 2016, 8, 3498–3506. [Google Scholar] [CrossRef]
- Clarke, T.L.; Mostoslavsky, R. DNA Repair as a Shared Hallmark in Cancer and Ageing. Mol. Oncol. 2022, 16, 3352–3379. [Google Scholar] [CrossRef] [PubMed]
- Alejandro Lagunas-Rangel, F.; María Bermúdez-Cruz, R. The Role of DNA Repair in Cellular Aging Process. In DNA Repair—An Update; IntechOpen: London, UK, 2019. [Google Scholar]
- Zorin, V.; Grekhova, A.; Pustovalova, M.; Zorina, A.; Smetanina, N.; Vorobyeva, N.; Kopnin, P.; Gilmutdinova, I.; Moskalev, A.; Osipov, A.N.; et al. Spontaneous γH2AX Foci in Human Dermal Fibroblasts in Relation to Proliferation Activity and Aging. Aging 2019, 11, 4536–4546. [Google Scholar] [CrossRef]
- Carrero, D.; Soria-Valles, C.; López-Otín, C. Hallmarks of Progeroid Syndromes: Lessons from Mice and Reprogrammed Cells. Dis. Model. Mech. 2016, 9, 719–735. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Abstract IA001: DNA Damage Repair: Impact on Aging and Cancer and Applications of Nutritional Interventions. Cancer Res. 2023, 83, IA001. [Google Scholar] [CrossRef]
- Rieckher, M.; Garinis, G.A.; Schumacher, B. Molecular Pathology of Rare Progeroid Diseases. Trends Mol. Med. 2021, 27, 907–922. [Google Scholar] [CrossRef]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The Plasma NAD + Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation Res. 2019, 22, 121–130. [Google Scholar] [CrossRef]
- McReynolds, M.R.; Chellappa, K.; Baur, J.A. Age-Related NAD+ Decline. Exp. Gerontol. 2020, 134, 110888. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Deng, X.; Yu, Y.; Luo, L.; Chen, X.; Zheng, J.; Qiu, Y.; Xiao, F.; Xie, X.; Zhao, Y.; et al. Association of Human Whole Blood NAD+ Contents With Aging. Front. Endocrinol. 2022, 13, 829658. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes With Key Roles in Inflammation. Front. Oncol. 2020, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Strømland, Ø.; Diab, J.; Ferrario, E.; Sverkeli, L.J.; Ziegler, M. The Balance between NAD+ Biosynthesis and Consumption in Ageing. Mech. Ageing Dev. 2021, 199, 111569. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Sedej, S.; Kroemer, G. NAD + Metabolism in Cardiac Health, Aging, and Disease. Circulation 2021, 144, 1795–1817. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shen, J.; Liu, C.; Kuang, Z.; Tang, Y.; Qian, Z.; Guan, M.; Yang, Y.; Zhan, Y.; Li, N.; et al. Nicotine Rebalances NAD+ Homeostasis and Improves Aging-Related Symptoms in Male Mice by Enhancing NAMPT Activity. Nat. Commun. 2023, 14, 900. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [PubMed]
- Vazquez-Villasenor, I.; Garwood, C.J.; Simpson, J.E.; Heath, P.R.; Mortiboys, H.; Wharton, S.B. Persistent DNA Damage Alters the Neuronal Transcriptome Suggesting Cell Cycle Dysregulation and Altered Mitochondrial Function. Eur. J. Neurosci. 2021, 54, 6987–7005. [Google Scholar] [CrossRef]
- Omari Shekaftik, S.; Nasirzadeh, N. 8-Hydroxy-2′-Deoxyguanosine (8-OHdG) as a Biomarker of Oxidative DNA Damage Induced by Occupational Exposure to Nanomaterials: A Systematic Review. Nanotoxicology 2021, 15, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Li, X.; Wang, R.; Yu, J.; Ye, M.; Mao, L.; Zhang, S.; Zheng, S. Association between Oxidative DNA Damage and Risk of Colorectal Cancer: Sensitive Determination of Urinary 8-Hydroxy-2′-Deoxyguanosine by UPLC-MS/MS Analysis. Sci. Rep. 2016, 6, 32581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent Impacts of Aging on Mitochondrial DNA Quantity and Quality in Humans. BMC Genom. 2017, 18, 890. [Google Scholar] [CrossRef]
- Knez, J.; Winckelmans, E.; Plusquin, M.; Thijs, L.; Cauwenberghs, N.; Gu, Y.; Staessen, J.A.; Nawrot, T.S.; Kuznetsova, T. Correlates of Peripheral Blood Mitochondrial DNA Content in a General Population. Am. J. Epidemiol. 2015, 183, 138–146. [Google Scholar] [CrossRef]
- Yang, L.; Lin, X.; Tang, H.; Fan, Y.; Zeng, S.; Jia, L.; Li, Y.; Shi, Y.; He, S.; Wang, H.; et al. Mitochondrial DNA Mutation Exacerbates Female Reproductive Aging via Impairment of the NADH/NAD+ Redox. Aging Cell 2020, 19, e13206. [Google Scholar] [CrossRef]
- Ziada, A.S.; Lu, M.Y.; Ignas-Menzies, J.; Paintsil, E.; Li, M.; Ogbuagu, O.; Saberi, S.; Hsieh, A.Y.Y.; Sattha, B.; Harrigan, P.R.; et al. Mitochondrial DNA Somatic Mutation Burden and Heteroplasmy Are Associated with Chronological Age, Smoking, and HIV Infection. Aging Cell 2019, 18, e13018. [Google Scholar] [CrossRef]
- Su, T.; Turnbull, D.; Greaves, L. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes 2018, 9, 182. [Google Scholar] [CrossRef]
- Zabihi Diba, L.; Mohaddes Ardebili, S.M.; Gharesouran, J.; Houshmand, M. Age-Related Decrease in MtDNA Content as a Consequence of MtDNA 4977 Bp Deletion. Mitochondrial DNA Part A 2016, 27, 3008–3012. [Google Scholar] [CrossRef]
- Bulgakova, O.; Kussainova, A.; Kakabayev, A.; Aripova, A.; Baikenova, G.; Izzotti, A.; Bersimbaev, R. The Level of Free-Circulating MtDNA in Patients with Radon-Induced Lung Cancer. Env. Res. 2022, 207, 112215. [Google Scholar] [CrossRef]
- Afrifa, J.; Zhao, T.; Yu, J. Circulating Mitochondria DNA, a Non-Invasive Cancer Diagnostic Biomarker Candidate. Mitochondrion 2019, 47, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Bulgakova, O.; Kausbekova, A.; Kussainova, A.; Kalibekov, N.; Serikbaiuly, D.; Bersimbaev, R. Involvement of Circulating Cell-Free Mitochondrial DNA and Proinflammatory Cytokines in Pathogenesis of Chronic Obstructive Pulmonary Disease and Lung Cancer. Asian Pac. J. Cancer Prev. 2021, 22, 1927–1933. [Google Scholar] [CrossRef]
- Mohd Khair, S.Z.N.; Abd Radzak, S.M.; Mohamed Yusoff, A.A. The Uprising of Mitochondrial DNA Biomarker in Cancer. Dis. Markers 2021, 2021, 7675269. [Google Scholar] [CrossRef]
- Nidadavolu, L.S.; Feger, D.; Chen, D.; Wu, Y.; Grodstein, F.; Gross, A.L.; Bennett, D.A.; Walston, J.D.; Oh, E.S.; Abadir, P.M. Associations between Circulating Cell-Free Mitochondrial DNA, Inflammatory Markers, and Cognitive and Physical Outcomes in Community Dwelling Older Adults. Immun. Ageing 2023, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Yang, J.; Guo, Y.; Liu, Y.; Zhong, X. Altered Levels of Circulating Mitochondrial DNA in Elderly People with Sarcopenia: Association with Mitochondrial Impairment. Exp. Gerontol. 2022, 163, 111802. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Westbrook, R.; Davalos, M.; Yang, H.; Walston, J.; Abadir, P. CIRCULATING CELL-FREE APOPTOTIC MITOCHONDRIAL DNA FRAGMENTS IN FRAIL OLD ADULTS. Innov. Aging 2018, 2, 61. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The Three ‘P’s of Mitophagy: PARKIN, PINK1, and Post-Translational Modifications. Genes. Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired Mitophagy Leads to Cigarette Smoke Stress-induced Cellular Senescence: Implications for Chronic Obstructive Pulmonary Disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Hua, F.; Fang, P.; Li, C.; Deng, F.; Chen, S.; Ying, J.; Wang, X. Regulation of Mitophagy by Sirtuin Family Proteins: A Vital Role in Aging and Age-Related Diseases. Front. Aging Neurosci. 2022, 14, 845330. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Liu, G.-H.; Qu, J. Mitochondrial Sirtuins, Metabolism, and Aging. J. Genet. Genom. 2022, 49, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD + in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Nikolajevic, J.; Ariaee, N.; Liew, A.; Abbasnia, S.; Fazeli, B.; Sabovic, M. The Role of MicroRNAs in Endothelial Cell Senescence. Cells 2022, 11, 1185. [Google Scholar] [CrossRef]
- Smith-Vikos, T.; Slack, F.J. MicroRNAs and Their Roles in Aging. J. Cell Sci. 2012, 125, 7–17. [Google Scholar] [CrossRef]
- Williams, J.; Smith, F.; Kumar, S.; Vijayan, M.; Reddy, P.H. Are MicroRNAs True Sensors of Ageing and Cellular Senescence? Ageing Res. Rev. 2017, 35, 350–363. [Google Scholar] [CrossRef]
- Faraonio, R.; Salerno, P.; Passaro, F.; Sedia, C.; Iaccio, A.; Bellelli, R.; Nappi, T.C.; Comegna, M.; Romano, S.; Salvatore, G.; et al. A Set of MiRNAs Participates in the Cellular Senescence Program in Human Diploid Fibroblasts. Cell Death Differ. 2012, 19, 713–721. [Google Scholar] [CrossRef]
- Munk, R.; Panda, A.C.; Grammatikakis, I.; Gorospe, M.; Abdelmohsen, K. Senescence-Associated MicroRNAs. Int. Rev. Cell Mol. Biol. 2017, 334, 177–205. [Google Scholar]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a Regulates Cardiac Ageing and Function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Maes, O.C.; Sarojini, H.; Wang, E. Stepwise Up-regulation of MicroRNA Expression Levels from Replicating to Reversible and Irreversible Growth Arrest States in WI-38 Human Fibroblasts. J. Cell Physiol. 2009, 221, 109–119. [Google Scholar] [CrossRef]
- Dellago, H.; Preschitz-Kammerhofer, B.; Terlecki-Zaniewicz, L.; Schreiner, C.; Fortschegger, K.; Chang, M.W.-F.; Hackl, M.; Monteforte, R.; Kühnel, H.; Schosserer, M.; et al. High Levels of OncomiR-21 Contribute to the Senescence-induced Growth Arrest in Normal Human Cells and Its Knock-down Increases the Replicative Lifespan. Aging Cell 2013, 12, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahè, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. MiR-146a Is Modulated in Human Endothelial Cell with Aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Wang, H.; Jia, C.; Zhu, S.; Chu, X.; Ma, Q.; Wei, J.; Chen, E.; Zhu, W.; Macon, C.J.; et al. MicroRNA-146a Induces Lineage-Negative Bone Marrow Cell Apoptosis and Senescence by Targeting Polo-Like Kinase 2 Expression. Arter. Thromb. Vasc. Biol. 2017, 37, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Cazalla, D.; Almstead, L.L.; Steitz, J.A.; DiMaio, D. MiR-29 and MiR-30 Regulate B-Myb Expression during Cellular Senescence. Proc. Natl. Acad. Sci. 2011, 108, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Takeshita, F.; Hino, Y.; Fukunaga, S.; Kudo, Y.; Tamaki, A.; Matsunaga, J.; Takahashi, R.; Takata, T.; Shimamoto, A.; et al. MiR-22 Represses Cancer Progression by Inducing Cellular Senescence. J. Cell Biol. 2011, 193, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; de Oliveira-Silva, T.; Lunardon, G.; Balbino-Silva, C.; Lima, V.M.; Huang, Z.-P.; Donato, J., Jr.; Takano, A.P.C.; Barreto-Chaves, M.L.; Wang, D.-Z.; et al. Ablation of MiRNA-22 Protects against Obesity-Induced Adipocyte Senescence and Ameliorates Metabolic Disorders in Middle-Aged Mice. Mech. Ageing Dev. 2023, 210, 111775. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.; Brunner, S.; Fortschegger, K.; Schreiner, C.; Micutkova, L.; Mück, C.; Laschober, G.T.; Lepperdinger, G.; Sampson, N.; Berger, P.; et al. MiR-17, MiR-19b, MiR-20a, and MiR-106a Are Down-regulated in Human Aging. Aging Cell 2010, 9, 291–296. [Google Scholar] [CrossRef] [PubMed]
- van Almen, G.C.; Verhesen, W.; van Leeuwen, R.E.W.; van de Vrie, M.; Eurlings, C.; Schellings, M.W.M.; Swinnen, M.; Cleutjens, J.P.M.; van Zandvoort, M.A.M.J.; Heymans, S.; et al. MicroRNA-18 and MicroRNA-19 Regulate CTGF and TSP-1 Expression in Age-related Heart Failure. Aging Cell 2011, 10, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Overhoff, M.G.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Bishop, C.L. Cellular Senescence Mediated by P16INK4A-Coupled MiRNA Pathways. Nucleic Acids Res. 2014, 42, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Ukai, T.; Sato, M.; Akutsu, H.; Umezawa, A.; Mochida, J. MicroRNA-199a-3p, MicroRNA-193b, and MicroRNA-320c Are Correlated to Aging and Regulate Human Cartilage Metabolism. J. Orthop. Res. 2012, 30, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Scheiber, M.N.; Neumann, C.; Calin, G.A.; Zhou, D. MicroRNA Regulation of Ionizing Radiation-Induced Premature Senescence. Endocrine 2011, 81, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Lai, M.; Chen, M.; Xie, C.; Liao, R.; Kang, Y.J.; Xiao, C.; Hu, W.-Y.; Han, J.; Sun, P. The MiR-17-92 Cluster of MicroRNAs Confers Tumorigenicity by Inhibiting Oncogene-Induced Senescence. Cancer Res. 2010, 70, 8547–8557. [Google Scholar] [CrossRef] [PubMed]
- Alsharif, G.; Jafri, I.; Berasa, R.; Ahmed, N.H.; Nunlee-Bland, G.; Fluitt, M.B.; Gambhir, K.K. Dysregulation of MiR144 and MiR451 Expression in the Circulating Human Erythrocytes from the African American Adults. ARC J. Diabetes Endocrinol. 2020, 6, 1–6. [Google Scholar] [CrossRef]
- Noureddine, S.; Nie, J.; Schneider, A.; Menon, V.; Fliesen, Z.; Dhahbi, J.; Victoria, B.; Oyer, J.; Robles-Carrillo, L.; Nunes, A.D.D.C.; et al. MicroRNA-449a Reduces Growth Hormone-Stimulated Senescent Cell Burden through PI3K-MTOR Signaling. Proc. Natl. Acad. Sci. USA 2023, 120, e2213207120. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative Stress Dependent MicroRNA-34a Activation via PI3Kα Reduces the Expression of Sirtuin-1 and Sirtuin-6 in Epithelial Cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Bei, Y.; Wu, X.; Cretoiu, D.; Shi, J.; Zhou, Q.; Lin, S.; Wang, H.; Cheng, Y.; Zhang, H.; Xiao, J.; et al. MiR-21 Suppression Prevents Cardiac Alterations Induced by d-Galactose and Doxorubicin. J. Mol. Cell Cardiol. 2018, 115, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Xue, M.; Xu, P.; Hu, F.; Sun, B.; Xiao, Z. MicroRNA Profiling Analysis Revealed Different Cellular Senescence Mechanisms in Human Mesenchymal Stem Cells Derived from Different Origin. Genomics 2017, 109, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, S.N.; Anderson, J.; Brown, T.J.; Nanus, D.E.; Housmans, B.; Green, J.A.; Hackl, M.; Choi, K.K.; Arkill, K.P.; Welting, T.; et al. The Role of Extracellular Vesicle MiRNAs and TRNAs in Synovial Fibroblast Senescence. Front. Mol. Biosci. 2022, 9, 971621. [Google Scholar] [CrossRef]
- Kern, F.; Kuhn, T.; Ludwig, N.; Simon, M.; Gröger, L.; Fabis, N.; Aparicio-Puerta, E.; Salhab, A.; Fehlmann, T.; Hahn, O.; et al. Ageing-Associated Small RNA Cargo of Extracellular Vesicles. RNA Biol. 2023, 20, 482–494. [Google Scholar] [CrossRef]
- Bogdanowicz, P.; Roullet, N.; Bensadoun, P.; Bessou-Touya, S.; Lemaitre, J.; Duplan, H. Reduction of Senescence-associated Secretory Phenotype and Exosome-shuttled miRNAs by Haritaki Fruit Extract in Senescent Dermal Fibroblasts. Int. J. Cosmet. Sci. 2023, 45, 488–499. [Google Scholar] [CrossRef]
- Farokhimanesh, S.; Komeili, A.; Nilforoushzadeh, M.A.; Zare, M. Inflamma-MiRs, Mito-MiRs, and SA-MiRs: Are They at the Crossroads of Inflammaging? J. Ski. Stem Cell 2018, 5, e81732. [Google Scholar] [CrossRef]
- Giuliani, A.; Micolucci, L.; Olivieri, F.; Procopio, A.D.; Rippo, M.R. MitomiRs in Human Inflamm-Aging. In Handbook of Immunosenescence; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–29. [Google Scholar]
- Rippo, M.R.; Olivieri, F.; Monsurrò, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in Human Inflamm-Aging: A Hypothesis Involving MiR-181a, MiR-34a and MiR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Liu, W.-Q.; Peng, J.-J.; Ma, Q.-L.; Peng, J.; Luo, X.-J. MiR-21-5p/203a-3p Promote Ox-LDL-Induced Endothelial Cell Senescence through down-Regulation of Mitochondrial Fission Protein Drp1. Mech. Ageing Dev. 2017, 164, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The MitomiR/Bcl-2 Axis Affects Mitochondrial Function and Autophagic Vacuole Formation in Senescent Endothelial Cells. Aging 2018, 10, 2855–2873. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.K.; Loucks, F.A.; Schroeder, E.K.; Bouchard, R.J.; Tyler, K.L.; Linseman, D.A. Glutathione Binding to the Bcl-2 Homology-3 Domain Groove. J. Biol. Chem. 2007, 282, 29296–29304. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Kubosumi, A.; Reddy, P.H. Mitochondrial MicroRNAs in Aging and Neurodegenerative Diseases. Cells 2020, 9, 1345. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Prattichizzo, F.; Micolucci, L.; Ceriello, A.; Procopio, A.D.; Rippo, M.R. Mitochondrial (Dys) Function in Inflammaging: Do MitomiRs Influence the Energetic, Oxidative, and Inflammatory Status of Senescent Cells? Mediat. Inflamm. 2017, 2017, 2309034. [Google Scholar] [CrossRef] [PubMed]
- la Torre, A.; Lo Vecchio, F.; Greco, A. Epigenetic Mechanisms of Aging and Aging-Associated Diseases. Cells 2023, 12, 1163. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Handy, D.E. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease (2013 Grover Conference Series). Pulm. Circ. 2014, 4, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, A.; Motevalizadeh Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P. Epigenetics: Principles and Practice. Dig. Dis. 2011, 29, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Hadad, N.; Masser, D.R.; Jackson, J.; Freeman, W.M.; Richardson, A. Revisiting the Genomic Hypomethylation Hypothesis of Aging. Ann. N. Y Acad. Sci. 2018, 1418, 69–79. [Google Scholar] [CrossRef]
- Sziráki, A.; Tyshkovskiy, A.; Gladyshev, V.N. Global Remodeling of the Mouse DNA Methylome during Aging and in Response to Calorie Restriction. Aging Cell 2018, 17, e12738. [Google Scholar] [CrossRef]
- So, A.-Y.; Jung, J.-W.; Lee, S.; Kim, H.-S.; Kang, K.-S. DNA Methyltransferase Controls Stem Cell Aging by Regulating BMI1 and EZH2 through MicroRNAs. PLoS ONE 2011, 6, e19503. [Google Scholar] [CrossRef]
- Nguyen, S.; Meletis, K.; Fu, D.; Jhaveri, S.; Jaenisch, R. Ablation of de Novo DNA Methyltransferase Dnmt3a in the Nervous System Leads to Neuromuscular Defects and Shortened Lifespan. Dev. Dyn. 2007, 236, 1663–1676. [Google Scholar] [CrossRef]
- Ciccarone, F.; Malavolta, M.; Calabrese, R.; Guastafierro, T.; Bacalini, M.G.; Reale, A.; Franceschi, C.; Capri, M.; Hervonen, A.; Hurme, M.; et al. Age-Dependent Expression of DNMT1 and DNMT3B in PBMCs from a Large European Population Enrolled in the MARK-AGE Study. Aging Cell 2016, 15, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Chow, M.Z.Y.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 Lysine 16 Hypoacetylation Is Associated with Defective DNA Repair and Premature Senescence in Zmpste24-Deficient Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Igata, T.; Etoh, K.; Koga, T.; Takebayashi, S.; Nakao, M. The NSD2/WHSC1/MMSET Methyltransferase Prevents Cellular Senescence-associated Epigenomic Remodeling. Aging Cell 2020, 19, e13173. [Google Scholar] [CrossRef] [PubMed]
- Persico, G.; Casciaro, F.; Amatori, S.; Rusin, M.; Cantatore, F.; Perna, A.; Auber, L.A.; Fanelli, M.; Giorgio, M. Histone H3 Lysine 4 and 27 Trimethylation Landscape of Human Alzheimer’s Disease. Cells 2022, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef]
- da Silva, P.F.L.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The Bystander Effect Contributes to the Accumulation of Senescent Cells in Vivo. Aging Cell 2019, 18, e12848. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Emerging Role of Extracellular Vesicles as a Senescence-Associated Secretory Phenotype: Insights into the Pathophysiology of Lung Diseases. Mol. Asp. Med. 2018, 60, 92–103. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Jakhar, R.; Crasta, K. Exosomes as Emerging Pro-Tumorigenic Mediators of the Senescence-Associated Secretory Phenotype. Int. J. Mol. Sci. 2019, 20, 2547. [Google Scholar] [CrossRef]
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small Extracellular Vesicles and Their MiRNA Cargo Are Anti-Apoptotic Members of the Senescence-Associated Secretory Phenotype. Aging 2018, 10, 1103–1132. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The Senescence-Associated Secretory Phenotype and Its Regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-KB) Signaling in Cancer Development and Immune Diseases. Genes. Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-ΚB by TNF Family Cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.; Carmody, R. The Regulation of NF-ΚB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Reale, C.; Iervolino, A.; Scudiero, I.; Ferravante, A.; D’Andrea, L.E.; Mazzone, P.; Zotti, T.; Leonardi, A.; Roberto, L.; Zannini, M.; et al. NF-ΚB Essential Modulator (NEMO) Is Critical for Thyroid Function. J. Biol. Chem. 2016, 291, 5765–5773. [Google Scholar] [CrossRef]
- Wang, W.; Mani, A.M.; Wu, Z.-H. DNA Damage-Induced Nuclear Factor-Kappa B Activation and Its Roles in Cancer Progression. J. Cancer Metastasis Treat. 2017, 3, 45. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, X.; Wang, S.; Mao, G. DNA Damage Response(DDR): A Link between Cellular Senescence and Human Cytomegalovirus. Virol. J. 2023, 20, 250. [Google Scholar] [CrossRef]
- Piret, B.; Schoonbroodt, S.; Piette, J. The ATM Protein Is Required for Sustained Activation of NF-ΚB Following DNA Damage. Oncogene 1999, 18, 2261–2271. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.U.; et al. ATM Is a Key Driver of NF-ΚB-Dependent DNA-Damage-Induced Senescence, Stem Cell Dysfunction and Aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The Senescent Bystander Effect Is Caused by ROS-Activated NF-ΚB Signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.-H.; Arany, P.R.; Huang, Y.-Y.; Tomkinson, E.M.; Sharma, S.K.; Kharkwal, G.B.; Saleem, T.; Mooney, D.; Yull, F.E.; Blackwell, T.S.; et al. Low-Level Laser Therapy Activates NF-KB via Generation of Reactive Oxygen Species in Mouse Embryonic Fibroblasts. PLoS ONE 2011, 6, e22453. [Google Scholar] [CrossRef] [PubMed]
- Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic Inflammation and the Hallmarks of Aging. Mol. Metab. 2023, 74, 101755. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and Aging: Signaling Pathways and Intervention Therapies. Signal Transduct. Target. Ther. 2023, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for Targeting Senescent Cells in Human Disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.; Li, X.-C.; Mi, W.-L.; Chu, Y.-X.; Wang, Y.-Q.; Mao-Ying, Q.-L. Neuronal Toll like Receptor 9 Contributes to Complete Freund’s Adjuvant-Induced Inflammatory Pain in Mice. Front. Mol. Neurosci. 2022, 15, 1008203. [Google Scholar] [CrossRef]
- Shen, J.; Dai, Z.; Li, Y.; Zhu, H.; Zhao, L. TLR9 Regulates NLRP3 Inflammasome Activation via the NF-KB Signaling Pathway in Diabetic Nephropathy. Diabetol. Metab. Syndr. 2022, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Mallavia, B.; Liu, F.; Lefrançais, E.; Cleary, S.J.; Kwaan, N.; Tian, J.J.; Magnen, M.; Sayah, D.M.; Soong, A.; Chen, J.; et al. Mitochondrial DNA Stimulates TLR9-Dependent Neutrophil Extracellular Trap Formation in Primary Graft Dysfunction. Am. J. Respir. Cell Mol. Biol. 2020, 62, 364–372. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.G.; Wenceslau, C.F.; Goulopoulou, S.; Ogbi, S.; Baban, B.; Sullivan, J.C.; Matsumoto, T.; Webb, R.C. Circulating Mitochondrial DNA and Toll-like Receptor 9 Are Associated with Vascular Dysfunction in Spontaneously Hypertensive Rats. Cardiovasc. Res. 2015, 107, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic Inflammation Induces Telomere Dysfunction and Accelerates Ageing in Mice. Nat. Commun. 2014, 5, 4172. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Martucci, M.; Chiariello, A.; Franceschi, C.; Salvioli, S. Mitochondria, Immunosenescence and Inflammaging: A Role for Mitokines? Semin. Immunopathol. 2020, 42, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kuk, M.U.; So, M.K.; Song, E.S.; Lee, H.; Ahn, S.K.; Kwon, H.W.; Park, J.T.; Park, S.C. Targeting Mitochondrial Oxidative Stress as a Strategy to Treat Aging and Age-Related Diseases. Antioxidants 2023, 12, 934. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial Reactive Oxygen Species Drive Proinflammatory Cytokine Production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Olivieri, F.; Prattichizzo, F.; Giuliani, A.; Matacchione, G.; Rippo, M.R.; Sabbatinelli, J.; Bonafè, M. MiR-21 and MiR-146a: The MicroRNAs of Inflammaging and Age-Related Diseases. Ageing Res. Rev. 2021, 70, 101374. [Google Scholar] [CrossRef]
- Liechty, C.; Hu, J.; Zhang, L.; Liechty, K.W.; Xu, J. Role of MicroRNA-21 and Its Underlying Mechanisms in Inflammatory Responses in Diabetic Wounds. Int. J. Mol. Sci. 2020, 21, 3328. [Google Scholar] [CrossRef]
- Liu, R.; Du, J.; Zhou, J.; Zhong, B.; Ba, L.; Zhang, J.; Liu, Y.; Liu, S. Elevated MicroRNA-21 Is a Brake of Inflammation Involved in the Development of Nasal Polyps. Front. Immunol. 2021, 12, 530488. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs MiR-146a/b Negatively Modulate the Senescence-Associated Inflammatory Mediators IL-6 and IL-8. Aging 2009, 1, 402–411. [Google Scholar] [CrossRef]
- Chan, B.D.; Wong, W.; Lee, M.M.; Cho, W.C.; Yee, B.K.; Kwan, Y.W.; Tai, W.C. Exosomes in Inflammation and Inflammatory Disease. Proteomics 2019, 19, e1800149. [Google Scholar] [CrossRef]
- Tong, J.; Hei, T.K. Aging and Age-Related Health Effects of Ionizing Radiation. Radiat. Med. Prot. 2020, 1, 15–23. [Google Scholar] [CrossRef]
- Li, P.; Hou, M.; Lou, F.; Björkholm, M.; Xu, D. Telomere Dysfunction Induced by Chemotherapeutic Agents and Radiation in Normal Human Cells. Int. J. Biochem. Cell Biol. 2012, 44, 1531–1540. [Google Scholar] [CrossRef]
- van de Kamp, G.; Heemskerk, T.; Kanaar, R.; Essers, J. DNA Double Strand Break Repair Pathways in Response to Different Types of Ionizing Radiation. Front. Genet. 2021, 12, 738230. [Google Scholar] [CrossRef]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of Ionizing Radiation on Biological Molecules—Mechanisms of Damage and Emerging Methods of Detection. Antioxid. Redox Signal 2014, 21, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Qi, F.; Kobayashi, J. Potential Relationship between the Biological Effects of Low-Dose Irradiation and Mitochondrial ROS Production. J. Radiat. Res. 2018, 59, ii91–ii97. [Google Scholar] [CrossRef] [PubMed]
- Krawczuk-Rybak, M.; Latoch, E. Risk Factors for Premature Aging in Childhood Cancer Survivors. Dev. Period. Med. 2019, 23, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Tabasso, A.F.S.; Jones, D.J.L.; Jones, G.D.D.; Macip, S. Radiotherapy-Induced Senescence and Its Effects on Responses to Treatment. Clin. Oncol. 2019, 31, 283–289. [Google Scholar] [CrossRef]
- Cupit-Link, M.C.; Kirkland, J.L.; Ness, K.K.; Armstrong, G.T.; Tchkonia, T.; LeBrasseur, N.K.; Armenian, S.H.; Ruddy, K.J.; Hashmi, S.K. Biology of Premature Ageing in Survivors of Cancer. ESMO Open 2017, 2, e000250. [Google Scholar] [CrossRef]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-Radiation Induced DNA Double-Strand Breaks: A Direct and Indirect Lighting Up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef]
- Imaoka, T.; Nishimura, M.; Daino, K.; Hosoki, A.; Takabatake, M.; Kokubo, T.; Doi, K.; Showler, K.; Nishimura, Y.; Moriyama, H.; et al. Age Modifies the Effect of 2-MeV Fast Neutrons on Rat Mammary Carcinogenesis. Radiat. Res. 2017, 188, 419. [Google Scholar] [CrossRef]
- Ulyanenko, S.; Pustovalova, M.; Koryakin, S.; Beketov, E.; Lychagin, A.; Ulyanenko, L.; Kaprin, A.; Grekhova, A.; Ozerova, A.M.; Ozerov, I.V.; et al. Formation of γH2AX and PATM Foci in Human Mesenchymal Stem Cells Exposed to Low Dose-Rate Gamma-Radiation. Int. J. Mol. Sci. 2019, 20, 2645. [Google Scholar] [CrossRef]
- Lowe, D.J.; Herzog, M.; Mosler, T.; Cohen, H.; Felton, S.; Beli, P.; Raj, K.; Galanty, Y.; Jackson, S.P. Chronic Irradiation of Human Cells Reduces Histone Levels and Deregulates Gene Expression. Sci. Rep. 2020, 10, 2200. [Google Scholar] [CrossRef]
- Noda, A. Radiation-Induced Unrepairable DSBs: Their Role in the Late Effects of Radiation and Possible Applications to Biodosimetry. J. Radiat. Res. 2018, 59, ii114–ii120. [Google Scholar] [CrossRef]
- Tang, F.R.; Liu, L.; Wang, H.; Ho, K.J.N.; Sethi, G. Spatiotemporal Dynamics of ΓH2AX in the Mouse Brain after Acute Irradiation at Different Postnatal Days with Special Reference to the Dentate Gyrus of the Hippocampus. Aging 2021, 13, 15815–15832. [Google Scholar] [CrossRef] [PubMed]
- Vaurijoux, A.; Voisin, P.; Freneau, A.; Barquinero, J.F.; Gruel, G. Transmission of Persistent Ionizing Radiation-Induced Foci through Cell Division in Human Primary Cells. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2017, 797–799, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Marková, E.; Torudd, J.; Belyaev, I. Long Time Persistence of Residual 53BP1/γ-H2AX Foci in Human Lymphocytes in Relationship to Apoptosis, Chromatin Condensation and Biological Dosimetry. Int. J. Radiat. Biol. 2011, 87, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, X.; Dong, Y.; Huo, Q.; Yue, T.; Wu, X.; Lu, L.; Zhang, J.; Zhao, Y.; Dong, H.; et al. Nicotinamide Riboside Intervention Alleviates Hematopoietic System Injury of Ionizing Radiation-induced Premature Aging Mice. Aging Cell 2023, 22, e13976. [Google Scholar] [CrossRef]
- Lee, J.T.; Gu, W. SIRT1: Regulator of P53 Deacetylation. Genes. Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Kobayashi, J.; Komatsu, K.; Kunugita, N. Severe Mitochondrial Damage Associated with Low-Dose Radiation Sensitivity in ATM- and NBS1-Deficient Cells. Cell Cycle 2016, 15, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, S.; Kashino, G.; Suzuki, K.; Yamashita, S.; Mori, H. Ionizing Radiation-Induced Cell Death Is Partly Caused by Increase of Mitochondrial Reactive Oxygen Species in Normal Human Fibroblast Cells. Radiat. Res. 2015, 183, 455. [Google Scholar] [CrossRef]
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.-C.; Gaugler, M.-H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing Radiation Induces Long-Term Senescence in Endothelial Cells through Mitochondrial Respiratory Complex II Dysfunction and Superoxide Generation. Free Radic. Biol. Med. 2017, 108, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing Radiation Induces Mitochondrial Reactive Oxygen Species Production Accompanied by Upregulation of Mitochondrial Electron Transport Chain Function and Mitochondrial Content under Control of the Cell Cycle Checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Zhu, F.; Deng, S.; Li, X.; He, Y. Regulatory Roles of MiR-22/Redd1-Mediated Mitochondrial ROS and Cellular Autophagy in Ionizing Radiation-Induced BMSC Injury. Cell Death Dis. 2019, 10, 227. [Google Scholar] [CrossRef]
- Szumiel, I. Ionizing Radiation-Induced Oxidative Stress, Epigenetic Changes and Genomic Instability: The Pivotal Role of Mitochondria. Int. J. Radiat. Biol. 2015, 91, 1–12. [Google Scholar] [CrossRef]
- Meyer, R.K.; Gilman, K.E.; Rheinheimer, B.A.; Meeks, L.; Limesand, K.H. AMPK Activation Restores Salivary Function Following Radiation Treatment. J. Dent. Res. 2023, 102, 546–554. [Google Scholar] [CrossRef]
- Liao, X.; Huang, X.; Li, X.; Qiu, X.; Li, M.; Liu, R.; He, T.; Tang, Q. AMPK Phosphorylates NAMPT to Regulate NAD + Homeostasis under Ionizing Radiation. Open Biol. 2022, 12, 220213. [Google Scholar] [CrossRef]
- Schauer, M.; Kottek, T.; Schönherr, M.; Bhattacharya, A.; Ibrahim, S.M.; Hirose, M.; Köhling, R.; Fuellen, G.; Schmitz, U.; Kunz, M. A Mutation in the NADH-Dehydrogenase Subunit 2 Suppresses Fibroblast Aging. Oncotarget 2015, 6, 8552–8566. [Google Scholar] [CrossRef] [PubMed]
- Prithivirajsingh, S.; Story, M.D.; Bergh, S.A.; Geara, F.B.; Kian Ang, K.; Ismail, S.M.; Stevens, C.W.; Buchholz, T.A.; Brock, W.A. Accumulation of the Common Mitochondrial DNA Deletion Induced by Ionizing Radiation. FEBS Lett. 2004, 571, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Antipova, V.N.; Lomaeva, M.G.; Zyrina, N.V. Mitochondrial DNA Deletions in Tissues of Mice after Ionizing Radiation Exposure. Int. J. Radiat. Biol. 2018, 94, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.-S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 Contributes to Repair of Mitochondrial DNA and Protects from Apoptotic Cell Death under Oxidative Stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [PubMed]
- Schilling-Tóth, B.; Sándor, N.; Kis, E.; Kadhim, M.; Sáfrány, G.; Hegyesi, H. Analysis of the Common Deletions in the Mitochondrial DNA Is a Sensitive Biomarker Detecting Direct and Non-Targeted Cellular Effects of Low Dose Ionizing Radiation. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 716, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, S.A.; Minkabirova, G.M.; Bezlepkin, V.G.; Gaziev, A.I. Cell-Free DNA in the Urine of Rats Exposed to Ionizing Radiation. Radiat. Env. Biophys. 2015, 54, 297–304. [Google Scholar] [CrossRef]
- Bisserier, M.; Shanmughapriya, S.; Rai, A.K.; Gonzalez, C.; Brojakowska, A.; Garikipati, V.N.S.; Madesh, M.; Mills, P.J.; Walsh, K.; Arakelyan, A.; et al. Cell-Free Mitochondrial DNA as a Potential Biomarker for Astronauts’ Health. J. Am. Heart Assoc. 2021, 10, e022055. [Google Scholar] [CrossRef]
- Ariyoshi, K.; Miura, T.; Kasai, K.; Fujishima, Y.; Nakata, A.; Yoshida, M. Radiation-Induced Bystander Effect Is Mediated by Mitochondrial DNA in Exosome-Like Vesicles. Sci. Rep. 2019, 9, 9103. [Google Scholar] [CrossRef]
- Yu, L.; Yang, X.; Li, X.; Qin, L.; Xu, W.; Cui, H.; Jia, Z.; He, Q.; Wang, Z. Pink1/PARK2/MROS-Dependent Mitophagy Initiates the Sensitization of Cancer Cells to Radiation. Oxid. Med. Cell Longev. 2021, 2021, 5595652. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Cheng, J.; Ding, X.; Yuan, Z. Phosphoglycerate Mutase 5-Mediated Mitochondrial Apoptosis/Necrosis Pathways and Mitophagy Determine the Mode of Cell Death Induced by Ionizing Radiation in Non-Small-Cell Lung Cancer. Endocrine 2016, 96, E584–E585. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, P.; Li, C.; Wang, W.; Zhang, T.; Li, J.; Li, H.; Dong, C.; Meng, W.; Zhou, H. Ionizing Radiation Triggers Mitophagy to Enhance DNA Damage in Cancer Cells. Cell Death Discov. 2023, 9, 267. [Google Scholar] [CrossRef]
- Pandey, B.N.; Gordon, D.M.; De Toledo, S.M.; Pain, D.; Azzam, E.I. Normal Human Fibroblasts Exposed to High- or Low-Dose Ionizing Radiation: Differential Effects on Mitochondrial Protein Import and Membrane Potential. Antioxid. Redox Signal 2006, 8, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zaharieva, E.K.; Sasatani, M.; Kobayashi, J. Possible Relationship between Mitochondrial Changes and Oxidative Stress under Low Dose-Rate Irradiation. Redox Rep. 2021, 26, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, V.; Aykin-Burns, N.; Tripathi, P.; Krager, K.J.; Sharma, S.K.; Moros, E.G.; Corry, P.M.; Nowak, G.; Hauer-Jensen, M.; Boerma, M. Radiation-Induced Alterations in Mitochondria of the Rat Heart. Radiat. Res. 2014, 181, 324. [Google Scholar] [CrossRef]
- Azimzadeh, O.; Azizova, T.; Merl-Pham, J.; Subramanian, V.; Bakshi, M.V.; Moseeva, M.; Zubkova, O.; Hauck, S.M.; Anastasov, N.; Atkinson, M.J.; et al. A Dose-Dependent Perturbation in Cardiac Energy Metabolism Is Linked to Radiation-Induced Ischemic Heart Disease in Mayak Nuclear Workers. Oncotarget 2017, 8, 9067–9078. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Abdalla, M.Y.; Moore, T.A.; Bartenhagen, L.; Case, A.J.; Zimmerman, M.C. Healthcare Workers Occupationally Exposed to Ionizing Radiation Exhibit Altered Levels of Inflammatory Cytokines and Redox Parameters. Antioxidants 2019, 8, 12. [Google Scholar] [CrossRef]
- Chen, B.; Dai, Q.; Zhang, Q.; Yan, P.; Wang, A.; Qu, L.; Jin, Y.; Zhang, D. The Relationship among Occupational Irradiation, DNA Methylation Status, and Oxidative Damage in Interventional Physicians. Medicine 2019, 98, e17373. [Google Scholar] [CrossRef]
- Jia, M.; Wang, Z. MicroRNAs as Biomarkers for Ionizing Radiation Injury. Front. Cell Dev. Biol. 2022, 10, 861451. [Google Scholar] [CrossRef]
- He, X.; Yang, A.; McDonald, D.G.; Riemer, E.C.; Vanek, K.N.; Schulte, B.A.; Wang, G.Y. MiR-34a Modulates Ionizing Radiation-Induced Senescence in Lung Cancer Cells. Oncotarget 2017, 8, 69797–69807. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Gandhi, M.; Kelly, L.; Nikiforov, Y.E. MicroRNA Dysregulation in Human Thyroid Cells Following Exposure to Ionizing Radiation. Thyroid 2011, 21, 261–266. [Google Scholar] [CrossRef]
- Liu, C.; Li, B.; Cheng, Y.; Lin, J.; Hao, J.; Zhang, S.; Mitchel, R.E.J.; Sun, D.; Ni, J.; Zhao, L.; et al. MiR-21 Plays an Important Role in Radiation Induced Carcinogenesis in BALB/c Mice by Directly Targeting the Tumor Suppressor Gene Big-H3. Int. J. Biol. Sci. 2011, 7, 347–363. [Google Scholar] [CrossRef]
- Chaudhry, M.A.; Omaruddin, R.A.; Brumbaugh, C.D.; Tariq, M.A.; Pourmand, N. Identification of Radiation-Induced MicroRNA Transcriptome by next-Generation Massively Parallel Sequencing. J. Radiat. Res. 2013, 54, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Huang, D.; Huang, X.; Wang, W.; Yang, S.; Chen, S. Exosomal MicroRNA-26b-5p Down-regulates ATF2 to Enhance Radiosensitivity of Lung Adenocarcinoma Cells. J. Cell Mol. Med. 2020, 24, 7730–7742. [Google Scholar] [CrossRef]
- Hu, Z.; Tie, Y.; Lv, G.; Zhu, J.; Fu, H.; Zheng, X. Transcriptional Activation of MiR-320a by ATF2, ELK1 and YY1 Induces Cancer Cell Apoptosis under Ionizing Radiation Conditions. Int. J. Oncol. 2018, 53, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, H.; Yu, J.; Xu, X.; Jing, H.; Li, N.; Tang, Y.; Wang, S.; Li, Y.; Cai, J.; et al. MiR-320b/RAD21 Axis Affects Hepatocellular Carcinoma Radiosensitivity to Ionizing Radiation Treatment through DNA Damage Repair Signaling. Cancer Sci. 2021, 112, 575–588. [Google Scholar] [CrossRef]
- Rogers, C.J.; Kyubwa, E.M.; Lukaszewicz, A.I.; Yamada-Hanff, J.; Starbird, M.A.; Miller, T.A.; Phelps, A.A.; Wallack, S.; Mahendra, S.; Thrall, K.; et al. Identification of MiRNA Associated with Reduced Survival after Whole-Thorax Lung Irradiation in Non-Human Primates. Radiat. Res. 2021, 196, 510–522. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, S.; Wen, Y.; Zhong, L. Effect of MicroRNA-210 on the Growth of Ovarian Cancer Cells and the Efficacy of Radiotherapy. Gynecol. Obs. Investig. 2021, 86, 71–80. [Google Scholar] [CrossRef]
- Jiang, P.; Rao, E.Y.; Meng, N.; Zhao, Y.; Wang, J.J. MicroRNA-17-92 Significantly Enhances Radioresistance in Human Mantle Cell Lymphoma Cells. Radiat. Oncol. 2010, 5, 100. [Google Scholar] [CrossRef]
- Menon, N.; Rogers, C.J.; Lukaszewicz, A.I.; Axtelle, J.; Yadav, M.; Song, F.; Chakravarti, A.; Jacob, N.K. Detection of Acute Radiation Sickness: A Feasibility Study in Non-Human Primates Circulating MiRNAs for Triage in Radiological Events. PLoS ONE 2016, 11, e0167333. [Google Scholar] [CrossRef]
- Templin, T.; Amundson, S.A.; Brenner, D.J.; Smilenov, L.B. Whole Mouse Blood MicroRNA as Biomarkers for Exposure to γ-Rays and 56Fe Ions. Int. J. Radiat. Biol. 2011, 87, 653–662. [Google Scholar] [CrossRef]
- Mao, A.; Zhao, Q.; Zhou, X.; Sun, C.; Si, J.; Zhou, R.; Gan, L.; Zhang, H. MicroRNA-449a Enhances Radiosensitivity by Downregulation of c-Myc in Prostate Cancer Cells. Sci. Rep. 2016, 6, 27346. [Google Scholar] [CrossRef]
- Summerer, I.; Unger, K.; Braselmann, H.; Schuettrumpf, L.; Maihoefer, C.; Baumeister, P.; Kirchner, T.; Niyazi, M.; Sage, E.; Specht, H.M.; et al. Circulating MicroRNAs as Prognostic Therapy Biomarkers in Head and Neck Cancer Patients. Br. J. Cancer 2015, 113, 76–82. [Google Scholar] [CrossRef]
- Chaudhry, M.A.; Omaruddin, R.A.; Kreger, B.; de Toledo, S.M.; Azzam, E.I. Micro RNA Responses to Chronic or Acute Exposures to Low Dose Ionizing Radiation. Mol. Biol. Rep. 2012, 39, 7549–7558. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, X.; Tang, X.; Wang, P.; Wang, H.; Wang, Y. MiR-21 Is Continually Elevated Long-Term in the Brain after Exposure to Ionizing Radiation. Radiat. Res. 2012, 177, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Kura, B.; Kalocayova, B.; LeBaron, T.W.; Frimmel, K.; Buday, J.; Surovy, J.; Slezak, J. Regulation of MicroRNAs by Molecular Hydrogen Contributes to the Prevention of Radiation-Induced Damage in the Rat Myocardium. Mol. Cell Biochem. 2019, 457, 61–72. [Google Scholar] [CrossRef]
- Gao, F.; Liu, P.; Narayanan, J.; Yang, M.; Fish, B.L.; Liu, Y.; Liang, M.; Jacobs, E.R.; Medhora, M. Changes in MiRNA in the Lung and Whole Blood after Whole Thorax Irradiation in Rats. Sci. Rep. 2017, 7, 44132. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, M.; Skoczylas, Ł.; Gawin, M.; Krzyżowska, M.; Pietrowska, M.; Widłak, P. Radiation-Induced Bystander Effect Mediated by Exosomes Involves the Replication Stress in Recipient Cells. Int. J. Mol. Sci. 2022, 23, 4169. [Google Scholar] [CrossRef]
- Xu, S.; Ding, N.; Pei, H.; Hu, W.; Wei, W.; Zhang, X.; Zhou, G.; Wang, J. MiR-21 Is Involved in Radiation-Induced Bystander Effects. RNA Biol. 2014, 11, 1161–1170. [Google Scholar] [CrossRef]
- Dinh, T.-K.T.; Fendler, W.; Chałubińska-Fendler, J.; Acharya, S.S.; O’Leary, C.; Deraska, P.V.; D’Andrea, A.D.; Chowdhury, D.; Kozono, D. Circulating MiR-29a and MiR-150 Correlate with Delivered Dose during Thoracic Radiation Therapy for Non-Small Cell Lung Cancer. Radiat. Oncol. 2016, 11, 61. [Google Scholar] [CrossRef]
- Kussainova, A.; Bulgakova, O.; Aripova, A.; Khalid, Z.; Bersimbaev, R.; Izzotti, A. The Role of Mitochondrial MiRNAs in the Development of Radon-Induced Lung Cancer. Biomedicines 2022, 10, 428. [Google Scholar] [CrossRef]
- Kumar, D.; Salian, S.R.; Kalthur, G.; Uppangala, S.; Kumari, S.; Challapalli, S.; Chandraguthi, S.G.; Krishnamurthy, H.; Jain, N.; Kumar, P.; et al. Semen Abnormalities, Sperm DNA Damage and Global Hypermethylation in Health Workers Occupationally Exposed to Ionizing Radiation. PLoS ONE 2013, 8, e69927. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Baulch, J.E. Epigenetic Changes and Nontargeted Radiation Effects—Is There a Link? Env. Mol. Mutagen. 2008, 49, 16–25. [Google Scholar] [CrossRef]
- Kamstra, J.H.; Hurem, S.; Martin, L.M.; Lindeman, L.C.; Legler, J.; Oughton, D.; Salbu, B.; Brede, D.A.; Lyche, J.L.; Aleström, P. Ionizing Radiation Induces Transgenerational Effects of DNA Methylation in Zebrafish. Sci. Rep. 2018, 8, 15373. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Nakamura, A.; Kovalchuk, O.; Koturbash, I.; Mitchell, S.A.; Marino, S.A.; Brenner, D.J.; Bonner, W.M. DNA Double-Strand Breaks Form in Bystander Cells after Microbeam Irradiation of Three-Dimensional Human Tissue Models. Cancer Res. 2007, 67, 4295–4302. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Rugo, R.E.; Hendricks, C.A.; Loree, J.; Thibault, B.; Kutanzi, K.; Pogribny, I.; Yanch, J.C.; Engelward, B.P.; Kovalchuk, O. Irradiation Induces DNA Damage and Modulates Epigenetic Effectors in Distant Bystander Tissue in Vivo. Oncogene 2006, 25, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Rugo, R.E.; Mutamba, J.T.; Mohan, K.N.; Yee, T.; Chaillet, J.R.; Greenberger, J.S.; Engelward, B.P. Methyltransferases Mediate Cell Memory of a Genotoxic Insult. Oncogene 2011, 30, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Miousse, I.R.; Sridharan, V.; Nzabarushimana, E.; Skinner, C.M.; Melnyk, S.B.; Pavliv, O.; Hauer-Jensen, M.; Nelson, G.A.; Boerma, M. Radiation-Induced Changes in DNA Methylation of Repetitive Elements in the Mouse Heart. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2016, 787, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.; Koturbash, I.; Tryndyak, V.; Hudson, D.; Stevenson, S.M.L.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Fractionated Low-Dose Radiation Exposure Leads to Accumulation of DNA Damage and Profound Alterations in DNA and Histone Methylation in the Murine Thymus. Mol. Cancer Res. 2005, 3, 553–561. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Xu, K.; Mao, X.; Xue, L.; Liu, X.; Yu, H.; Chen, L.; Chu, X. Genome-Wide Screen of DNA Methylation Changes Induced by Low Dose X-Ray Radiation in Mice. PLoS ONE 2014, 9, e90804. [Google Scholar] [CrossRef]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and Histone H4 Acetylation at Lysine 16 Are Critical for DNA Damage Response and Double-Strand Break Repair. Mol. Cell Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef]
- Bhadra, M.P.; Horikoshi, N.; Pushpavallipvalli, S.N.; Sarkar, A.; Bag, I.; Krishnan, A.; Lucchesi, J.C.; Kumar, R.; Yang, Q.; Pandita, R.K.; et al. The Role of MOF in the Ionizing Radiation Response Is Conserved in Drosophila Melanogaster. Chromosoma 2012, 121, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Neumayer, G.; Nguyen, M.D. TPX2 Impacts Acetylation of Histone H4 at Lysine 16: Implications for DNA Damage Response. PLoS ONE 2014, 9, e110994. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone Demethylase KDM5B Is a Key Regulator of Genome Stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef]
- Seiler, D.M.; Rouquette, J.; Schmid, V.J.; Strickfaden, H.; Ottmann, C.; Drexler, G.A.; Mazurek, B.; Greubel, C.; Hable, V.; Dollinger, G.; et al. Double-Strand Break-Induced Transcriptional Silencing Is Associated with Loss of Tri-Methylation at H3K4. Chromosome Res. 2011, 19, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Di Nisio, E.; Lupo, G.; Licursi, V.; Negri, R. The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation. Front. Genet. 2021, 12, 639602. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.H.; Waung, I.; Camphausen, K.; Tofilon, P.J. Inhibition of the Histone H3K27 Demethylase UTX Enhances Tumor Cell Radiosensitivity. Mol. Cancer Ther. 2018, 17, 1070–1078. [Google Scholar] [CrossRef]
- Bueno, M.T.D.; Baldascini, M.; Richard, S.; Lowndes, N.F. Recruitment of Lysine Demethylase 2A to DNA Double Strand Breaks and Its Interaction with 53BP1 Ensures Genome Stability. Oncotarget 2018, 9, 15915–15930. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Elbakrawy, E.; Kaur Bains, S.; Bright, S.; AL-Abedi, R.; Mayah, A.; Goodwin, E.; Kadhim, M. Radiation-Induced Senescence Bystander Effect: The Role of Exosomes. Biology 2020, 9, 191. [Google Scholar] [CrossRef]
- Al-Mayah, A.H.J.; Irons, S.L.; Pink, R.C.; Carter, D.R.F.; Kadhim, M.A. Possible Role of Exosomes Containing RNA in Mediating Nontargeted Effect of Ionizing Radiation. Radiat. Res. 2012, 177, 539–545. [Google Scholar] [CrossRef]
- Kumar Jella, K.; Rani, S.; O’Driscoll, L.; McClean, B.; Byrne, H.J.; Lyng, F.M. Exosomes Are Involved in Mediating Radiation Induced Bystander Signaling in Human Keratinocyte Cells. Radiat. Res. 2014, 181, 138–145. [Google Scholar] [CrossRef]
- Kawai, H.; Yamada, Y.; Tatsuka, M.; Niwa, O.; Yamamoto, K.; Suzuki, F. Down-Regulation of Nuclear Factor KappaB Is Required for P53-Dependent Apoptosis in X-Ray-Irradiated Mouse Lymphoma Cells and Thymocytes. Cancer Res. 1999, 59, 6038–6041. [Google Scholar] [PubMed]
- Jonak, K.; Kurpas, M.; Szoltysek, K.; Janus, P.; Abramowicz, A.; Puszynski, K. A Novel Mathematical Model of ATM/P53/NF- ΚB Pathways Points to the Importance of the DDR Switch-off Mechanisms. BMC Syst. Biol. 2016, 10, 75. [Google Scholar] [CrossRef]
- Dong, X.-R.; Luo, M.; Fan, L.; Zhang, T.; Liu, L.; Dong, J.-H.; Wu, G. Corilagin Inhibits the Double Strand Break-Triggered NF-KappaB Pathway in Irradiated Microglial Cells. Int. J. Mol. Med. 2010, 25, 531–536. [Google Scholar]
- Bock, F.J.; Peintner, L.; Tanzer, M.; Manzl, C.; Villunger, A. P53-Induced Protein with a Death Domain (PIDD): Master of Puppets? Oncogene 2012, 31, 4733–4739. [Google Scholar] [CrossRef]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD Mediates NF-ΚB Activation in Response to DNA Damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef]
- Zając, G.; Rusin, M.; Łasut-Szyszka, B.; Puszyński, K.; Widłak, P. Activation of the Atypical NF-ΚB Pathway Induced by Ionizing Radiation Is Not Affected by the P53 Status. Acta Biochim. Pol. 2022, 69, 205–210. [Google Scholar] [CrossRef]
- Li, N.; Karin, M. Ionizing Radiation and Short Wavelength UV Activate NF-ΚB through Two Distinct Mechanisms. Proc. Natl. Acad. Sci. USA 1998, 95, 13012–13017. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Dimtchev, A.; Lavin, M.F.; Dritschilo, A.; Jung, M. A Novel Ionizing Radiation-Induced Signaling Pathway That Activates the Transcription Factor NF-ΚB. Oncogene 1998, 17, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Ong, Z.Y.; Gibson, R.J.; Bowen, J.M.; Stringer, A.M.; Darby, J.M.; Logan, R.M.; Yeoh, A.S.; Keefe, D.M. Pro-Inflammatory Cytokines Play a Key Role in the Development of Radiotherapy-Induced Gastrointestinal Mucositis. Radiat. Oncol. 2010, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Yu, T.; Chen, G.; Brown, S.A.; Yu, Z.; Mattson, M.P.; Thompson, J.S. Effects of NF-ΚB1 (P50) Targeted Gene Disruption on Ionizing Radiation-Induced NF-ΚB Activation and TNFα, IL-1α, IL-1β and IL-6 MRNA Expression in Vivo. Int. J. Radiat. Biol. 2001, 77, 763–772. [Google Scholar] [CrossRef]
- Song, Y.; Okazaki, R.; Yoshida, Y. Senescence-Associated Secretory Phenotype and Activation of NF-ΚB in Splenocytes of Old Mice Exposed to Irradiation at a Young Age. Dev. Comp. Immunol. 2021, 122, 104124. [Google Scholar] [CrossRef] [PubMed]
- Baselet, B.; Belmans, N.; Coninx, E.; Lowe, D.; Janssen, A.; Michaux, A.; Tabury, K.; Raj, K.; Quintens, R.; Benotmane, M.A.; et al. Functional Gene Analysis Reveals Cell Cycle Changes and Inflammation in Endothelial Cells Irradiated with a Single X-Ray Dose. Front. Pharmacol. 2017, 8, 253802. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meeren, A.; Bertho, J.-M.; Vandamme, M.; Gaugler, M.-H. Ionizing Radiation Enhances IL-6 and IL-8 Production by Human Endothelial Cells. Mediat. Inflamm. 1997, 6, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Azimzadeh, O.; Sievert, W.; Sarioglu, H.; Merl-Pham, J.; Yentrapalli, R.; Bakshi, M.V.; Janik, D.; Ueffing, M.; Atkinson, M.J.; Multhoff, G.; et al. Integrative Proteomics and Targeted Transcriptomics Analyses in Cardiac Endothelial Cells Unravel Mechanisms of Long-Term Radiation-Induced Vascular Dysfunction. J. Proteome Res. 2015, 14, 1203–1219. [Google Scholar] [CrossRef]
- Singh, V.; Gupta, D.; Arora, R. NF-ΚB as a Key Player in Regulation of Cellular Radiation Responses and Identification of Radiation Countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef]
- Janus, P.; Szołtysek, K.; Zając, G.; Stokowy, T.; Walaszczyk, A.; Widłak, W.; Wojtaś, B.; Gielniewski, B.; Iwanaszko, M.; Braun, R.; et al. Pro-Inflammatory Cytokine and High Doses of Ionizing Radiation Have Similar Effects on the Expression of NF-KappaB-Dependent Genes. Cell Signal 2018, 46, 23–31. [Google Scholar] [CrossRef]
- Johnston, C.J.; Piedboeuf, B.; Rubin, P.; Williams, J.P.; Baggs, R.; Finkelstein, J.N. Early and Persistent Alterations in the Expression of Interleukin-1 Alpha, Interleukin-1 Beta and Tumor Necrosis Factor Alpha MRNA Levels in Fibrosis-Resistant and Sensitive Mice after Thoracic Irradiation. Radiat. Res. 1996, 145, 762–767. [Google Scholar] [CrossRef]
- Kim, S.; Choe, J.H.; Lee, G.J.; Kim, Y.S.; Kim, S.Y.; Lee, H.-M.; Jin, H.S.; Kim, T.S.; Kim, J.-M.; Cho, M.-J.; et al. Ionizing Radiation Induces Innate Immune Responses in Macrophages by Generation of Mitochondrial Reactive Oxygen Species. Radiat. Res. 2016, 187, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, S.; Guan, H.; Zhou, P.-K. Radiation-Induced Non-Targeted Effect of Immunity Provoked by Mitochondrial DNA Damage Triggered CGAS/ AIM2 Pathways. Radiat. Med. Prot. 2022, 3, 47–55. [Google Scholar] [CrossRef]
- LoBianco, F.V.; Krager, K.J.; Carter, G.S.; Alam, S.; Yuan, Y.; Lavoie, E.G.; Dranoff, J.A.; Aykin-Burns, N. The Role of Sirtuin 3 in Radiation-Induced Long-Term Persistent Liver Injury. Antioxidants 2020, 9, 409. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, Y.; Du, L.; Xu, C.; Cao, J.; Fan, T.; Liu, J.; Su, X.; Fan, S.; Liu, Q.; et al. Resveratrol Inhibits Ionising Irradiation-Induced Inflammation in MSCs by Activating SIRT1 and Limiting NLRP-3 Inflammasome Activation. Int. J. Mol. Sci. 2013, 14, 14105–14118. [Google Scholar] [CrossRef] [PubMed]
- Calveley, V.L.; Khan, M.A.; Yeung, I.W.T.; Vandyk, J.; Hill, R.P. Partial Volume Rat Lung Irradiation: Temporal Fluctuations of in-Field and out-of-Field DNA Damage and Inflammatory Cytokines Following Irradiation. Int. J. Radiat. Biol. 2005, 81, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Morishita, Y.; Khattree, R.; Misumi, M.; Sasaki, K.; Hayashi, I.; Yoshida, K.; Kajimura, J.; Kyoizumi, S.; Imai, K.; et al. Evaluation of Systemic Markers of Inflammation in Atomic-bomb Survivors with Special Reference to Radiation and Age Effects. FASEB J. 2012, 26, 4765–4773. [Google Scholar] [CrossRef]
- Scoggin, C.H. The Cellular Basis of Aging. West. J. Med. 1981, 135, 521–525. [Google Scholar]
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718. [Google Scholar] [CrossRef]
- van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular Senescence: A Key Therapeutic Target in Aging and Diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| miRNAs | Expression | Effect | Ref. |
|---|---|---|---|
| miR-34a | ↑ | Targets SIRT1 and reduces its expression, leading to the activation of p53 and induction of cellular senescence | [83] |
| It is actively expressed in cardiac cells of aged mice, and inhibition or deletion reduces age-associated cardiomyocyte death | [84] | ||
| Increased expression in H2O2-induced premature cellular senescence | [85] | ||
| miR-21 | ↑ | Overexpression reduces the replicative lifespan of HUVECs | [86] |
| miR-146a | ↓ | Reduced expression in senescent HUVECs | [87] |
| ↑ | Increased expression of senescent lin-BMCs | [88] | |
| miR-29 | ↑ | Rb-dependent increase in expression during replication and induced cellular senescence | [89] |
| miR-22 | ↑ | Promotes cellular senescence in human fibroblasts and epithelial cells by targeting CDK6, Sp1, and SIRT1 | [90] |
| Ablation prevented cellular senescence in white adipose tissue (WAT) induced by obesity | [91] | ||
| miR-19b | ↓ | Six models of cellular senescence show reduced expression | [92] |
| Reduced expression in age-associated cardiac aging | [93] | ||
| miR-26b | ↑ | Overexpression induces cellular senescence in human epithelial cells and fibroblasts | [94] |
| miR-320c | ↓ | Decreased expression with age and participation in juvenile chondrocyte properties by regulating ADAMTS5 expression | [95] |
| miR-199a-3p | ↑ | Increased expression with age; involvement in chondrocyte senescence by regulating aggrecan, COL2, and SOX9 | [95] |
| miR-20a | ↓ | Six models of cellular senescence show reduced expression | [92] |
| Overexpression inhibits stress-induced senescence in WI-38 cells | [96] | ||
| miR-155 | ↓ | Reduced expression in Ras-induced senescent WI-38 cells; knockdown increases stress-induced cellular senescence | [96] |
| miR-210 | ↑ | Overexpression induces senescence in human epithelial cells and fibroblasts | [94] |
| Induces the formation of γH2AX foci and ROS in IMR90 cells; increased expression results in an age-related phenotype | [82] | ||
| miR-106a | ↓ | Five models of cellular senescence show reduced expression | [92] |
| miR-17-92 | ↓ | Increased expression in primary human fibroblasts inhibits Ras-induced cellular senescence | [97] |
| miR-15a | ↓ | Involved in the regulation of stress-induced senescence of WI-38 cells | [96] |
| miR-144 | ↑ | Increased expression in aged erythrocytes from type 2 diabetic patients | [98] |
| miR-494 | ↑ | Overexpression enhances DNA damage and cellular senescence in IMR90 cells | [82] |
| miR-449a | ↓ | Increased expression slows senescence in HUVECs and adipose tissue by targeting p16Ink4a, p21CIP1, and the PI3K-mTOR signaling pathway | [99] |
| miR-17 | ↓ | All seven models of cellular senescence show reduced expression | [92] |
| miR-25 | ↓ | Downregulation in Ras-induced senescent WI-38 cells | [96] |
| miR-431 | ↑ | Increased expression in both replicative and stress-induced senescent human lung fibroblasts | [96] |
| Cell Type | Radiation | Study Design | Effect of γH2AX | Ref. |
|---|---|---|---|---|
| Neurons | γ-rays 5 Gy | Whole-body irradiation of mice at postnatal day 3 (P3), P10, and P21. Animals were euthanized at 1, 7, and 120 day(s) and 15 months after irradiation | Consistently demonstrated radiation-induced γH2AX foci or PDDF, in the brains of mice at 120 days and 15 months after irradiation at P3, P10, and P21 | [181] |
| Lymphocytes | γ-rays 0.5–10 Gy | Human G(0)-lymphocytes were irradiated at doses ranging from 0.5 to 10 Gy. The dose response of γH2AX foci was analyzed 24 h, 96 h, 1 week, 2 weeks, and 4 weeks after irradiation | Residual γH2AX foci persisted in human lymphocytes up to 4 weeks after irradiation | [183] |
| HUVECs | X-rays 1 or 5 Gy | Synchronized G0/G1 phase HUVECs were irradiated with an X-ray dose of 1 or 5 Gy and IRIF were studied from 10 min to 7 days after irradiation | By 7 days after irradiation with 5 Gy, the mean number of γH2AX IRIF per nucleus was still significantly more than in unirradiated cells | [182] |
| miRNAs | Effect | Ref. |
|---|---|---|
| miR-34a | Targeting c-Myc suppressed its expression, which markedly enhanced IR-induced senescence in human NSCLC cells | [212] |
| ↑ expression was more than two-fold in thyroid cells shortly after IR irradiation | [213] | |
| Expression reached a maximum at 3 days in WI-38 cells after exposure to IR, participating in the induction of senescence, and by day 7, the expression level decreased to an insignificant level | [96] | |
| miR-21 | ↑ expression levels in radiation-induced thymic lymphoma tissue samples in BALB/c mice | [214] |
| miR-146a | ↑ expression levels at 8 h and 24 h in TK6 cells exposed to X-rays | [215] |
| miR-29a-3p | ↑ expression in exosomes during IR-induced fibroblast senescence | [105] |
| miR-22 | ↑ expression in rBMSCs after IR irradiation enhanced mtROS production and reduced cell viability | [190] |
| miR-19b | ↓ expression only in IR-induced senescence in WI-38 cells | [96] |
| miR-26b-5p | ↓ expression in blood serum is associated with poor survival in patients with lung adenocarcinoma after radiotherapy | [216] |
| miR-320a | Linear ↑ in expression in HeLa cells as a function of IR dose and treatment duration | [217] |
| miR-320b | ↓ expression attenuated IR-treatment-induced DNA damage in HCC cells | [218] |
| miR-199a-3p | Expression in plasma at or below the median abundance at day 6 was associated with decreased survival and early mortality after irradiation in non-human primates (NHP) | [219] |
| miR-20a | ↓ expression levels in IR-induced senescence in WI-38 cells | [96] |
| miR-155 | ↓ expression levels in IR-induced senescence in WI-38 cells | [96] |
| miR-210 | Overexpression in OVCAR3 and SKOV3 cancer cell lines reduced their sensitivity to radiotherapy after IR irradiation. | [220] |
| miR-106a | ↓ expression levels in both IR-induced and replicative senescence in WI-38 cells | [96] |
| miR-17-92 | Overexpression significantly increased survival of Z138c cells after IR treatment | [221] |
| miR-15a | ↓ expression levels in IR-induced senescence in WI-38 cells | [96] |
| miR-144 | ↑ expression in plasma of irradiated animals on days 3 and/or 7 | [222] |
| miR-494 | Differential expression in the blood of mice irradiated Fe56 | [223] |
| miR-449a | ↑ expression in IR action of LNCaP cells | [224] |
| miR-17 | ↓ expression levels in both IR-induced and replicative senescence in WI-38 cells | [96] |
| miR-25 | ↓ expression levels in IR-induced senescence in WI-38 cells | [96] |
| miR-431 | ↑ expression levels in IR-induced senescence in WI-38 cells | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibragimova, M.; Kussainova, A.; Aripova, A.; Bersimbaev, R.; Bulgakova, O. The Molecular Mechanisms in Senescent Cells Induced by Natural Aging and Ionizing Radiation. Cells 2024, 13, 550. https://doi.org/10.3390/cells13060550
Ibragimova M, Kussainova A, Aripova A, Bersimbaev R, Bulgakova O. The Molecular Mechanisms in Senescent Cells Induced by Natural Aging and Ionizing Radiation. Cells. 2024; 13(6):550. https://doi.org/10.3390/cells13060550
Chicago/Turabian StyleIbragimova, Milana, Assiya Kussainova, Akmaral Aripova, Rakhmetkazhi Bersimbaev, and Olga Bulgakova. 2024. "The Molecular Mechanisms in Senescent Cells Induced by Natural Aging and Ionizing Radiation" Cells 13, no. 6: 550. https://doi.org/10.3390/cells13060550