
EZH2-Myc Hallmark in Oncovirus/Cytomegalovirus Infections and Cytomegalovirus’ Resemblance to Oncoviruses

Abstract
:1. Introduction
2. EZH2 and Myc: Key Players in Cancers
3. Activation of EZH2-Myc Axis by Oncoviruses
4. Activation of EZH2-Myc Axis by HCMV
5. Impact of EZH2 and Myc on Immunity
6. Oncoviruses and HCMV Foster a Pro-Oncogenic Environment in the Presence of EZH2 and Myc
7. Comparative Oncogenic Traits: HCMV-Resembling Oncoviruses
8. Conclusive Thoughts
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATL | Adult T-cell leukemia |
| Arg-1 | Arginase-1 |
| ARID1A | AT-rich interaction domain 1A |
| AURKA | Aurora kinase A |
| BARTs | Bam-HI A rightward transcripts |
| BR | Basic region |
| BIN1 | Bridging integrator 1 |
| BRD4 | Bromodomain-4 |
| CAK | Cdk-activating kinase |
| CEGBCs | CMV-elicited glioblastoma cells |
| CTH | CMV-transformed HMECs |
| CTO cells | CMV-transformed OECs |
| E2F1 | E2 promoter binding factor 1 |
| EBER1 | EBV-encoded RNAs |
| EBNAs | EBV nuclear antigens |
| EED | Embryonic ectoderm development |
| (EZH2 | Enhancer of zeste homolog 2 |
| EMT | Epithelial–mesenchymal transition |
| EBV | Epstein–Barr virus |
| FAO | Fatty acid oxidation |
| FOXM1 | Forkhead box protein M1 |
| GBM | Glioblastoma |
| GSCs | Glioblastoma stem cells |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| HLH–LZ | Helix–loop–helix–leucine zipper |
| HBV | Hepatitis B virus |
| HBx | Hepatitis B virus X protein |
| HCV | Hepatitis C virus |
| HGSOC | High-grade serous ovarian carcinoma |
| HR | High-risk |
| HR-HPV | High-risk human papillomavirus |
| HBZ | HTLV-1 bZIP factor |
| HCMV | Human cytomegalovirus |
| HIV | Human immunodeficiency virus |
| HMECs | Human mammary epithelial cells |
| HPV | Human papillomavirus |
| HTLV-1 | Human T-cell lymphotropic virus |
| hTERT | Human telomerase reverse transcriptase |
| HIF1A | Hypoxia-inducible-factor 1A |
| iNOS | Inducible nitric oxide synthase |
| ICAM1 | Intercellular adhesion molecule 1 |
| IFN | Interferon |
| IFNGR1 | Interferon-γ receptor 1 |
| IL-6 | Interlukin-6 |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| KS | Kaposi’s sarcoma |
| KO | Knockout |
| LT | Large T |
| LANA | Latency-associated nuclear antigen |
| LMP1 | Latent membrane protein 1 |
| LMPs | Latent membrane proteins |
| lncRNAs | Long non-coding RNAs |
| LR | Low-risk |
| MHC I | Major histocompatibility complex class I |
| MMPs | Matrix metalloproteinases |
| MED1 | Mediator 1 |
| MCC | Merkel cell carcinoma |
| MCPyV | Merkel cell polyomavirus |
| MCV | Merkel cell polyomavirus |
| LT | Large T |
| MICA | MHC class I polypeptide–related sequence A |
| miRNAs | MicroRNAs |
| MS | Mitochondrial superoxide |
| MAPK | Mitogen-activated protein kinase |
| E boxes | Myc binding sites |
| MBs | Myc homology boxes |
| MAX | Myc-associated factor X |
| Miz-1 | Myc-interacting zinc-finger protein-1 |
| MDSCs | Myeloid-derived suppressor cells |
| NK | Natural killer |
| NKG2D | Natural-killer group 2, member D |
| Nos2 | Nitric oxide synthase 2 |
| ncRNAs | Non-coding RNAs |
| NFκB | Nuclear factor kappa B |
| OECs | Ovarian epithelial cells |
| PBMCs | Peripheral blood mononuclear cells |
| PTEN | Phosphatase and tensin homolog |
| pRb | Phosphorylated retinoblastoma |
| PI3K | Phosphotylinosital 3 kinase |
| Akt | Protein kinase B |
| PBRM1 | Polybromo-1 |
| PcGs | Polycomb group genes |
| PRC2 | Polycomb repressive complex 2 |
| PGCCs | Polyploid giant cancer cells |
| P-TEFb | Positive transcription elongation factor b |
| PD-L1 | Programmed death-ligand 1 |
| PLZF | Promyelocytic leukemia zinc finger protein |
| (PMT | Proneural mesenchymal transition |
| PPIs | Protein–protein interactions |
| ROS | Reactive oxygen species |
| Tregs | Regulatory T cells |
| Rb | Retinoblastoma |
| RORα | Retinoic acid receptor-related orphan receptor alpha |
| SOX2 | Sex-determining region Y-box 2, also known as |
| SCFFBW7 | SKP1–cullin-1–F-box complex that contains FBW7 as the F-box protein |
| SP1 | Specificity protein 1 |
| SEC | Super elongation complex |
| SMARCA4 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4 |
| Th17 | T helper 17 |
| TBP | TATA box-binding protein |
| TA | Telomerase activity |
| TAD | Transactivation domain |
| TRRAP | Transformation/transcription domain-associated protein |
| TGF-β | Transforming growth factor beta |
| TNBC | Triple-negative breast cancer |
| TME | Tumor microenvironment |
| TNF | Tumor necrosis factor |
| TAMs | Tumor-associated macrophages |
| ULBP1 | UL16 binding protein 1 |
| VEGF | Vascular endothelial growth factor |
| vFLIP | Viral FLICE-inhibitory protein |
| vIRF3 | Viral IFN regulatory factor-3 |
| vIL-10 | Viral interlukin-10 |
| WDR5 | WD repeat-containing protein 5 |
| YY1 | Yin Yang 1 |
References
- Schiller, J.T.; Lowy, D.R. An Introduction to Virus Infections and Human Cancer. Viruses Hum. Cancer 2021, 217, 1–11. [Google Scholar] [CrossRef]
- Mui, U.; Haley, C.; Tyring, S. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef]
- Herbein, G. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses 2018, 10, 408. [Google Scholar] [CrossRef]
- Herbein, G. High-Risk Oncogenic Human Cytomegalovirus. Viruses 2022, 14, 2462. [Google Scholar] [CrossRef] [PubMed]
- Kachuri, L.; Francis, S.S.; Morrison, M.L.; Wendt, G.A.; Bossé, Y.; Cavazos, T.B.; Rashkin, S.R.; Ziv, E.; Witte, J.S. The Landscape of Host Genetic Factors Involved in Immune Response to Common Viral Infections. Genome Med. 2020, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Krump, N.A.; You, J. Molecular Mechanisms of Viral Oncogenesis in Humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Strumillo, S.T.; Kartavykh, D.; De Carvalho, F.F.; Cruz, N.C.; De Souza Teodoro, A.C.; Sobhie Diaz, R.; Curcio, M.F. Host–Virus Interaction and Viral Evasion. Cell Biol. Int. 2021, 45, 1124–1147. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Guven-Maiorov, E.; Tsai, C.-J.; Nussinov, R. Oncoviruses Can Drive Cancer by Rewiring Signaling Pathways Through Interface Mimicry. Front. Oncol. 2019, 9, 1236. [Google Scholar] [CrossRef]
- El Baba, R.; Herbein, G. Immune Landscape of CMV Infection in Cancer Patients: From “Canonical” Diseases toward Virus-Elicited Oncomodulation. Front. Immunol. 2021, 12, 730765. [Google Scholar] [CrossRef]
- Nehme, Z.; Pasquereau, S.; Haidar Ahmad, S.; El Baba, R.; Herbein, G. Polyploid Giant Cancer Cells, EZH2 and Myc Upregulation in Mammary Epithelial Cells Infected with High-Risk Human Cytomegalovirus. eBioMedicine 2022, 80, 104056. [Google Scholar] [CrossRef]
- El Baba, R.; Pasquereau, S.; Haidar Ahmad, S.; Monnien, F.; Abad, M.; Bibeau, F.; Herbein, G. EZH2-Myc Driven Glioblastoma Elicited by Cytomegalovirus Infection of Human Astrocytes. Oncogene 2023, 42, 2031–2045. [Google Scholar] [CrossRef]
- El Baba, R.; Haidar Ahmad, S.; Monnien, F.; Mansar, R.; Bibeau, F.; Herbein, G. Polyploidy, EZH2 Upregulation, and Transformation in Cytomegalovirus-Infected Human Ovarian Epithelial Cells. Oncogene 2023, 42, 3047–3061. [Google Scholar] [CrossRef] [PubMed]
- Bouezzedine, F.; El Baba, R.; Haidar Ahmad, S.; Herbein, G. Polyploid Giant Cancer Cells Generated from Human Cytomegalovirus-Infected Prostate Epithelial Cells. Cancers 2023, 15, 4994. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, C.; Song, Z.; Wang, H.; Ye, M.; Wang, D.; Kang, W.; Liu, H.; Qing, G. EZH2 Depletion Potentiates MYC Degradation Inhibiting Neuroblastoma and Small Cell Carcinoma Tumor Formation. Nat. Commun. 2022, 13, 12. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q. The Roles of EZH2 in Cancer and Its Inhibitors. Med. Oncol. 2023, 40, 167. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic Targeting of “Undruggable” MYC. eBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene—The Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Sun, J.; Cai, X.; Yung, M.M.; Zhou, W.; Li, J.; Zhang, Y.; Li, Z.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S.; et al. miR-137 Mediates the Functional Link between c-Myc and EZH2 That Regulates Cisplatin Resistance in Ovarian Cancer. Oncogene 2019, 38, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Paskeh, M.D.A.; Mehrabi, A.; Gholami, M.H.; Zabolian, A.; Ranjbar, E.; Saleki, H.; Ranjbar, A.; Hashemi, M.; Ertas, Y.N.; Hushmandi, K.; et al. EZH2 as a New Therapeutic Target in Brain Tumors: Molecular Landscape, Therapeutic Targeting and Future Prospects. Biomed. Pharmacother. 2022, 146, 112532. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 Histone Methyltransferase in Cancer Epigenetics. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in Cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Li, B.; Chng, W.-J. EZH2 Abnormalities in Lymphoid Malignancies: Underlying Mechanisms and Therapeutic Implications. J. Hematol. Oncol. 2019, 12, 118. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.-W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; DuPrie, M.L.; Krueger, H.; Merajver, S.D.; Ventura, A.C.; Toy, K.A.; Kleer, C.G. Histone Methyltransferase EZH2 Induces Akt-Dependent Genomic Instability and BRCA1 Inhibition in Breast Cancer. Cancer Res. 2011, 71, 2360–2370. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-Mutant Cancers Depend on Catalytic and Non-Catalytic Activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef]
- Yi, C.; Li, G.; Wang, W.; Sun, Y.; Zhang, Y.; Zhong, C.; Stovall, D.B.; Li, D.; Shi, J.; Sui, G. Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development. Cancers 2021, 13, 2402. [Google Scholar] [CrossRef]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC Protein Interactors in Gene Transcription and Cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative Approaches to Target Myc for Cancer Treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic Activity of the C-Myc Protein Requires Dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A Helix-Loop-Helix Zipper Protein That Forms a Sequence-Specific DNA-Binding Complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef]
- Helander, S.; Montecchio, M.; Pilstål, R.; Su, Y.; Kuruvilla, J.; Elvén, M.; Ziauddin, J.M.E.; Anandapadamanaban, M.; Cristobal, S.; Lundström, P.; et al. Pre-Anchoring of Pin1 to Unphosphorylated c-Myc in a Fuzzy Complex Regulates c-Myc Activity. Structure 2015, 23, 2267–2279. [Google Scholar] [CrossRef]
- Wang, J.; Yu, X.; Gong, W.; Liu, X.; Park, K.-S.; Ma, A.; Tsai, Y.-H.; Shen, Y.; Onikubo, T.; Pi, W.-C.; et al. EZH2 Noncanonically Binds cMyc and P300 through a Cryptic Transactivation Domain to Mediate Gene Activation and Promote Oncogenesis. Nat. Cell Biol. 2022, 24, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Role of Virus-Induced Host Cell Epigenetic Changes in Cancer. Int. J. Mol. Sci. 2021, 22, 8346. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, S.; Pei, M.; Zhao, M.; Wang, L.; Jiang, Y.; Yang, T.; Zhao, J.; Song, L.; Yang, X. Crosstalk between Histone Modification and DNA Methylation Orchestrates the Epigenetic Regulation of the Costimulatory Factors, Tim-3 and Galectin-9, in Cervical Cancer. Oncol. Rep. 2019, 42, 2655–2669. [Google Scholar] [CrossRef]
- Fujikawa, D.; Nakagawa, S.; Hori, M.; Kurokawa, N.; Soejima, A.; Nakano, K.; Yamochi, T.; Nakashima, M.; Kobayashi, S.; Tanaka, Y.; et al. Polycomb-Dependent Epigenetic Landscape in Adult T-Cell Leukemia. Blood 2016, 127, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, W.; Bakken, T.; Schutten, M.; Toth, Z.; Jung, J.U.; Gill, P.; Cannon, M.; Gao, S.-J. Cancer Angiogenesis Induced by Kaposi Sarcoma–Associated Herpesvirus Is Mediated by EZH2. Cancer Res. 2012, 72, 3582–3592. [Google Scholar] [CrossRef]
- Li, M.; Damania, B.; Alvarez, X.; Ogryzko, V.; Ozato, K.; Jung, J.U. Inhibition of P300 Histone Acetyltransferase by Viral Interferon Regulatory Factor. Mol. Cell. Biol. 2000, 20, 8254–8263. [Google Scholar] [CrossRef]
- Wei, X.; Xiang, T.; Ren, G.; Tan, C.; Liu, R.; Xu, X.; Wu, Z. miR-101 Is down-Regulated by the Hepatitis B Virus x Protein and Induces Aberrant DNA Methylation by Targeting DNA Methyltransferase 3A. Cell. Signal. 2013, 25, 439–446. [Google Scholar] [CrossRef]
- Classon, M.; Harlow, E. The Retinoblastoma Tumour Suppressor in Development and Cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef]
- Nair, J.S.; Ho, A.L.; Tse, A.N.; Coward, J.; Cheema, H.; Ambrosini, G.; Keen, N.; Schwartz, G.K. Aurora B Kinase Regulates the Postmitotic Endoreduplication Checkpoint via Phosphorylation of the Retinoblastoma Protein at Serine 780. MBoC 2009, 20, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Liao, Y.-J.; Cai, M.-Y.; Liu, Y.-H.; Liu, T.-H.; Chen, S.-P.; Bian, X.-W.; Guan, X.-Y.; Lin, M.C.; Zeng, Y.-X.; et al. The Putative Tumour Suppressor microRNA-124 Modulates Hepatocellular Carcinoma Cell Aggressiveness by Repressing ROCK2 and EZH2. Gut 2012, 61, 278–289. [Google Scholar] [CrossRef]
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3, e00478-18. [Google Scholar] [CrossRef] [PubMed]
- Khattri, M.; Amako, Y.; Gibbs, J.R.; Collura, J.L.; Arora, R.; Harold, A.; Li, M.Y.; Harms, P.W.; Ezhkova, E.; Shuda, M. Methyltransferase-Independent Function of Enhancer of Zeste Homologue 2 Maintains Tumorigenicity Induced by Human Oncogenic Papillomavirus and Polyomavirus. Tumour Virus Res. 2023, 16, 200264. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, Y.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Dömling, A.; et al. Survivin Is a Therapeutic Target in Merkel Cell Carcinoma. Sci. Transl. Med. 2012, 4, 133ra56. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.; Saltarella, I.; Vinella, A.; Muzio, L.; Pannone, G.; Fumarulo, R.; Vacca, A.; Mariggi, M. Survivin Overexpression in Head and Neck Squamous Cell Carcinomas as a New Therapeutic Target (Review). Oncol. Rep. 2019, 41, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-Dependent Merkel Cell Carcinoma to a Differentiated Neuron-like Phenotype by T Antigen Inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 20104–20114. [Google Scholar] [CrossRef]
- Bracken, A.P. EZH2 Is Downstream of the pRB-E2F Pathway, Essential for Proliferation and Amplified in Cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and Cell Cycle Control. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Liu, X.; Disbrow, G.L.; Yuan, H.; Tomaić, V.; Schlegel, R. Myc and Human Papillomavirus Type 16 E7 Genes Cooperate To Immortalize Human Keratinocytes. J. Virol. 2007, 81, 12689–12695. [Google Scholar] [CrossRef]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human Papillomavirus E6 and Myc Proteins Associate in Vivo and Bind to and Cooperatively Activate the Telomerase Reverse Transcriptase Promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef]
- Park, A.; Oh, S.; Jung, K.L.; Choi, U.Y.; Lee, H.-R.; Rosenfeld, M.G.; Jung, J.U. Global Epigenomic Analysis of KSHV-Infected Primary Effusion Lymphoma Identifies Functional MYC Superenhancers and Enhancer RNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 21618–21627. [Google Scholar] [CrossRef]
- Beer, S.; Wange, L.E.; Zhang, X.; Kuklik-Roos, C.; Enard, W.; Hammerschmidt, W.; Scialdone, A.; Kempkes, B. EBNA2-EBF1 Complexes Promote MYC Expression and Metabolic Processes Driving S-Phase Progression of Epstein-Barr Virus–Infected B Cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2200512119. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, J.; Wang, L.; Huang, S.; Chen, J. Human T-Cell Lymphotropic Virus Type 1 and Its Oncogenesis. Acta Pharmacol. Sin. 2017, 38, 1093–1103. [Google Scholar] [CrossRef]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.-M. Hepatitis C Virus-Induced Activation of β-Catenin Promotes c-Myc Expression and a Cascade of pro-Carcinogenetic Events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef]
- Lee, S.; Kim, W.; Ko, C.; Ryu, W.-S. Hepatitis B Virus X Protein Enhances Myc Stability by Inhibiting SCFSkp2 Ubiquitin E3 Ligase-Mediated Myc Ubiquitination and Contributes to Oncogenesis. Oncogene 2016, 35, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Klucky, B.; Wintersberger, E. Polyomavirus Small T Antigen Transactivates Genes by Its Ability to Provoke the Synthesis and the Stabilization of MYC. Oncogene 2007, 26, 6356–6360. [Google Scholar] [CrossRef] [PubMed]
- Prochownik, E.V.; Li, Y. The Ever Expanding Role for C-Myc in Promoting Genomic Instability. Cell Cycle 2007, 6, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Nehme, Z. Polyploid Giant Cancer Cells, a Hallmark of Oncoviruses and a New Therapeutic Challenge. Front. Oncol. 2020, 10, 567116. [Google Scholar] [CrossRef]
- Mahara, S.; Chng, W.J.; Yu, Q. Molecular Switch of EZH2 in Hypoxia. Cell Cycle 2016, 15, 3007–3008. [Google Scholar] [CrossRef] [PubMed]
- Nehme, Z.; Pasquereau, S.; Haidar Ahmad, S.; Coaquette, A.; Molimard, C.; Monnien, F.; Algros, M.-P.; Adotevi, O.; Diab Assaf, M.; Feugeas, J.-P.; et al. Polyploid Giant Cancer Cells, Stemness and Epithelial-Mesenchymal Plasticity Elicited by Human Cytomegalovirus. Oncogene 2021, 40, 3030–3046. [Google Scholar] [CrossRef]
- Li, Q.; Dang, C.V. C-Myc Overexpression Uncouples DNA Replication from Mitosis. Mol. Cell. Biol. 1999, 19, 5339–5351. [Google Scholar] [CrossRef]
- Kinouchi, T.; Saiki, S.; Naoe, T.; Uenaka, A.; Kotake, T.; Shiku, H.; Nakayama, E. Correlation of C-Myc Expression with Nuclear Pleomorphism in Human Renal Cell Carcinoma. Cancer Res. 1989, 49, 3627–3630. [Google Scholar]
- Kumar, A.; Tripathy, M.K.; Pasquereau, S.; Al Moussawi, F.; Abbas, W.; Coquard, L.; Khan, K.A.; Russo, L.; Algros, M.-P.; Valmary-Degano, S.; et al. The Human Cytomegalovirus Strain DB Activates Oncogenic Pathways in Mammary Epithelial Cells. EBioMedicine 2018, 30, 167–183. [Google Scholar] [CrossRef]
- El Baba, R.; Pasquereau, S.; Haidar Ahmad, S.; Diab-Assaf, M.; Herbein, G. Oncogenic and Stemness Signatures of the High-Risk HCMV Strains in Breast Cancer Progression. Cancers 2022, 14, 4271. [Google Scholar] [CrossRef]
- Eich, M.-L.; Athar, M.; Ferguson, J.E.; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, J.; Guo, Z.; Li, C.; Tan, Z.; Wang, J.; Yang, J.; Xue, L. Easy or Not—The Advances of EZH2 in Regulating T Cell Development, Differentiation, and Activation in Antitumor Immunity. Front. Immunol. 2021, 12, 741302. [Google Scholar] [CrossRef] [PubMed]
- Stairiker, C.J.; Thomas, G.D.; Salek-Ardakani, S. EZH2 as a Regulator of CD8+ T Cell Fate and Function. Front. Immunol. 2020, 11, 593203. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, A.V.; Mutis, T.; Roemer, M.G.M.; Scheijen, B.; Chamuleau, M.E.D. Impact of MYC on Anti-Tumor Immune Responses in Aggressive B Cell Non-Hodgkin Lymphomas: Consequences for Cancer Immunotherapy. Cancers 2020, 12, 3052. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, J.N.; Wang, R. MYC in Regulating Immunity: Metabolism and Beyond. Genes 2017, 8, 88. [Google Scholar] [CrossRef]
- Wang, X.; Brea, L.T.; Yu, J. Immune Modulatory Functions of EZH2 in the Tumor Microenvironment: Implications in Cancer Immunotherapy. Am. J. Clin. Exp. Urol. 2019, 7, 85–91. [Google Scholar]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.M.; Liou, Y.C.; Yu, Q. Context-Specific Regulation of NF-κB Target Gene Expression by EZH2 in Breast Cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Wee, Z.N.; Li, Z.; Lee, P.L.; Lee, S.T.; Lim, Y.P.; Yu, Q. EZH2-Mediated Inactivation of IFN-γ-JAK-STAT1 Signaling Is an Effective Therapeutic Target in MYC-Driven Prostate Cancer. Cell Rep. 2014, 8, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Gupta, R.; Green, M.R.; Wajapeyee, N. EZH2 Inhibits NK Cell–Mediated Antitumor Immunity by Suppressing CXCL10 Expression in an HDAC10-Dependent Manner. Proc. Natl. Acad. Sci. USA 2021, 118, e2102718118. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, T.; Wu, Z.; Zhu, D.; Gu, H. The Effects of MYC on Tumor Immunity and Immunotherapy. Cell Death Discov. 2023, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt Signaling in Breast Cancer: Biological Mechanisms, Challenges and Opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and Mechanisms of WNT Pathway Tumour Suppressors in Cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC Regulates the Antitumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Meškytė, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. [Google Scholar] [CrossRef] [PubMed]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I.; et al. Role of C-MYC in Alternative Activation of Human Macrophages and Tumor-Associated Macrophage Biology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Long, X.; Zhang, L.; Ye, Y.; Guo, J.; Liu, P.; Zhang, R.; Ning, J.; Yu, W.; Wei, F.; et al. Neurotensin/IL-8 Pathway Orchestrates Local Inflammatory Response and Tumor Invasion by Inducing M2 Polarization of Tumor-Associated Macrophages and Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma Cells. OncoImmunology 2018, 7, e1440166. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting Tumor-Associated Macrophages to Synergize Tumor Immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Hadjidaniel, M.D.; Muthugounder, S.; Hung, L.T.; Sheard, M.A.; Shirinbak, S.; Chan, R.Y.; Nakata, R.; Borriello, L.; Malvar, J.; Kennedy, R.J.; et al. Tumor-Associated Macrophages Promote Neuroblastoma via STAT3 Phosphorylation and up-Regulation of c-MYC. Oncotarget 2017, 8, 91516–91529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, H.; Zhao, S.; Wang, E.; Zhu, J.; Feng, D.; Zhu, Y.; Dou, W.; Fan, Q.; Hu, J.; et al. Epigenetic Silencing of miR-144/451a Cluster Contributes to HCC Progression via Paracrine HGF/MIF-Mediated TAM Remodeling. Mol. Cancer 2021, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhu, X.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 Enhances Expression of CCL5 to Promote Recruitment of Macrophages and Invasion in Lung Cancer. Biotechnol. Appl. Biochem. 2020, 67, 1011–1019. [Google Scholar] [CrossRef]
- Kang, N.; Eccleston, M.; Clermont, P.-L.; Latarani, M.; Male, D.K.; Wang, Y.; Crea, F. EZH2 Inhibition: A Promising Strategy to Prevent Cancer Immune Editing. Epigenomics 2020, 12, 1457–1476. [Google Scholar] [CrossRef]
- Qi, B.; Yang, C.; Zhu, Z.; Chen, H. EZH2-Inhibited MicroRNA-454-3p Promotes M2 Macrophage Polarization in Glioma. Front. Cell Dev. Biol. 2020, 8, 574940. [Google Scholar] [CrossRef]
- Yin, H.; Wang, Y.; Wu, Y.; Zhang, X.; Zhang, X.; Liu, J.; Wang, T.; Fan, J.; Sun, J.; Yang, A.; et al. EZH2-Mediated Epigenetic Silencing of miR-29/miR-30 Targets LOXL4 and Contributes to Tumorigenesis, Metastasis, and Immune Microenvironment Remodeling in Breast Cancer. Theranostics 2020, 10, 8494–8512. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yu, F.; Xu, D.; Zheng, H.; Li, M. EZH2, a Prominent Orchestrator of Genetic and Epigenetic Regulation of Solid Tumor Microenvironment and Immunotherapy. Biochim. Biophys. Acta BBA Rev. Cancer 2022, 1877, 188700. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 78, 653–669.e8. [Google Scholar] [CrossRef] [PubMed]
- Müller-Coan, B.G.; Caetano, B.F.R.; Pagano, J.S.; Elgui De Oliveira, D. Cancer Progression Goes Viral: The Role of Oncoviruses in Aggressiveness of Malignancies. Trends Cancer 2018, 4, 485–498. [Google Scholar] [CrossRef]
- Huang, D.; Song, S.-J.; Wu, Z.-Z.; Wu, W.; Cui, X.-Y.; Chen, J.-N.; Zeng, M.-S.; Su, S.-C. Epstein–Barr Virus-Induced VEGF and GM-CSF Drive Nasopharyngeal Carcinoma Metastasis via Recruitment and Activation of Macrophages. Cancer Res. 2017, 77, 3591–3604. [Google Scholar] [CrossRef]
- Lechien, J.R.; Descamps, G.; Seminerio, I.; Furgiuele, S.; Dequanter, D.; Mouawad, F.; Badoual, C.; Journe, F.; Saussez, S. HPV Involvement in the Tumor Microenvironment and Immune Treatment in Head and Neck Squamous Cell Carcinomas. Cancers 2020, 12, 1060. [Google Scholar] [CrossRef]
- Davis, R.J.; Van Waes, C.; Allen, C.T. Overcoming Barriers to Effective Immunotherapy: MDSCs, TAMs, and Tregs as Mediators of the Immunosuppressive Microenvironment in Head and Neck Cancer. Oral Oncol. 2016, 58, 59–70. [Google Scholar] [CrossRef]
- Sakakibara, S.; Tosato, G. Viral Interleukin-6: Role in Kaposi’s Sarcoma-Associated Herpesvirus–Associated Malignancies. J. Interferon Cytokine Res. 2011, 31, 791–801. [Google Scholar] [CrossRef]
- Karabajakian, A.; Ray-Coquard, I.; Blay, J.-Y. Molecular Mechanisms of Kaposi Sarcoma Development. Cancers 2022, 14, 1869. [Google Scholar] [CrossRef]
- Broussard, G.; Damania, B. KSHV: Immune Modulation and Immunotherapy. Front. Immunol. 2020, 10, 3084. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Da Silva, S.R.; Qin, H.; Ferris, R.L.; Tan, R.; Chang, Y.; Moore, P.S. The Central Repeat Domain 1 of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Latency Associated-Nuclear Antigen 1 (LANA1) Prevents Cis MHC Class I Peptide Presentation. Virology 2011, 412, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Shi, Y.; Zhang, M.; Goswami, S.; Afridi, S.; Meng, L.; Ma, J.; Chen, Y.; Lin, Y.; Zhang, J.; et al. Global Immune Characterization of HBV/HCV-Related Hepatocellular Carcinoma Identifies Macrophage and T-Cell Subsets Associated with Disease Progression. Cell Discov. 2020, 6, 90. [Google Scholar] [CrossRef]
- Nakahata, S.; Enriquez-Vera, D.; Jahan, M.I.; Sugata, K.; Satou, Y. Understanding the Immunopathology of HTLV-1-Associated Adult T-Cell Leukemia/Lymphoma: A Comprehensive Review. Biomolecules 2023, 13, 1543. [Google Scholar] [CrossRef] [PubMed]
- Schlemeyer, T.; Ohnezeit, D.; Virdi, S.; Körner, C.; Weißelberg, S.; Starzonek, S.; Schumacher, U.; Grundhoff, A.; Indenbirken, D.; Albertini, S.; et al. Merkel Cell Carcinoma and Immune Evasion: Merkel Cell Polyomavirus Small T-Antigen–Induced Surface Changes Can Be Reverted by Therapeutic Intervention. J. Investig. Dermatol. 2022, 142, 3071–3081.e13. [Google Scholar] [CrossRef]
- Mitteldorf, C.; Berisha, A.; Tronnier, M.; Pfaltz, M.C.; Kempf, W. PD-1 and PD-L1 in Neoplastic Cells and the Tumor Microenvironment of Merkel Cell Carcinoma. J. Cutan. Pathol. 2017, 44, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Haidar Ahmad, S.; El Baba, R.; Herbein, G. Polyploid Giant Cancer Cells, Cytokines and Cytomegalovirus in Breast Cancer Progression. Cancer Cell Int. 2023, 23, 119. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G. Tumors and Cytomegalovirus: An Intimate Interplay. Viruses 2022, 14, 812. [Google Scholar] [CrossRef]
- Khan, K.A.; Coaquette, A.; Davrinche, C.; Herbein, G. Bcl-3-Regulated Transcription from Major Immediate-Early Promoter of Human Cytomegalovirus in Monocyte-Derived Macrophages. J. Immunol. 2009, 182, 7784–7794. [Google Scholar] [CrossRef]
- Denaro, N.; Merlano, M.C.; Lo Nigro, C. Long Noncoding RNAs as Regulators of Cancer Immunity. Mol. Oncol. 2019, 13, 61–73. [Google Scholar] [CrossRef]
- Tai-Schmiedel, J.; Karniely, S.; Lau, B.; Ezra, A.; Eliyahu, E.; Nachshon, A.; Kerr, K.; Suárez, N.; Schwartz, M.; Davison, A.J.; et al. Human Cytomegalovirus Long Noncoding RNA4.9 Regulates Viral DNA Replication. PLoS Pathog. 2020, 16, e1008390. [Google Scholar] [CrossRef] [PubMed]
- Moussawi, F.A.; Kumar, A.; Pasquereau, S.; Tripathy, M.K.; Karam, W.; Diab-Assaf, M.; Herbein, G. The Transcriptome of Human Mammary Epithelial Cells Infected with the HCMV-DB Strain Displays Oncogenic Traits. Sci. Rep. 2018, 8, 12574. [Google Scholar] [CrossRef] [PubMed]
- Haidar Ahmad, S.; Pasquereau, S.; El Baba, R.; Nehme, Z.; Lewandowski, C.; Herbein, G. Distinct Oncogenic Transcriptomes in Human Mammary Epithelial Cells Infected with Cytomegalovirus. Front. Immunol. 2021, 12, 772160. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| EZH2/PRC2 Inhibitors | Myc Inhibitors | ||
|---|---|---|---|
| Inhibitor Name | Target | Inhibitor Name | Target |
| Tazemetostat, EL1, GSK126, CPI-169, EPZ005687, EPZ011989, ZLD10A, GSK503, JQEZ5, GSK926, GSK343, PF-06726304, MS1943, EZH2-IN-3, CPI-1205, EBI-2511, UNC1999, valemetostat, (R)-OR-S1, PF-06821497, oxetinib (AZD9291) | EZH2 | QN-1, APTO-253, AZD5153, GSK525762, dBET1 | Myc transcription |
| MLN0128, silvestrol, eFT226, BTYNB | Myc translation | ||
| Pyrimidine, derivatives, SZL-P1–41, TD19, volasertib | Myc stability | ||
| MYCMI-6, KI-MS2-008, Omomyc, FPPa-OmoMYC | Myc-Max heterodimer | ||
| PROTAC EED Degrader-1, PROTAC EED Degrader-2, UNC6852 | EED | Sulfopin, ASH2L-derived peptides, C620-0696 | Accessibility of Myc to downstream genes |
| Viral Pathogen | EZH2 | Myc | ||
|---|---|---|---|---|
| Involved Oncogene(s) | EZH2 Interactions | Involved Oncogene(s) | Myc Interactions | |
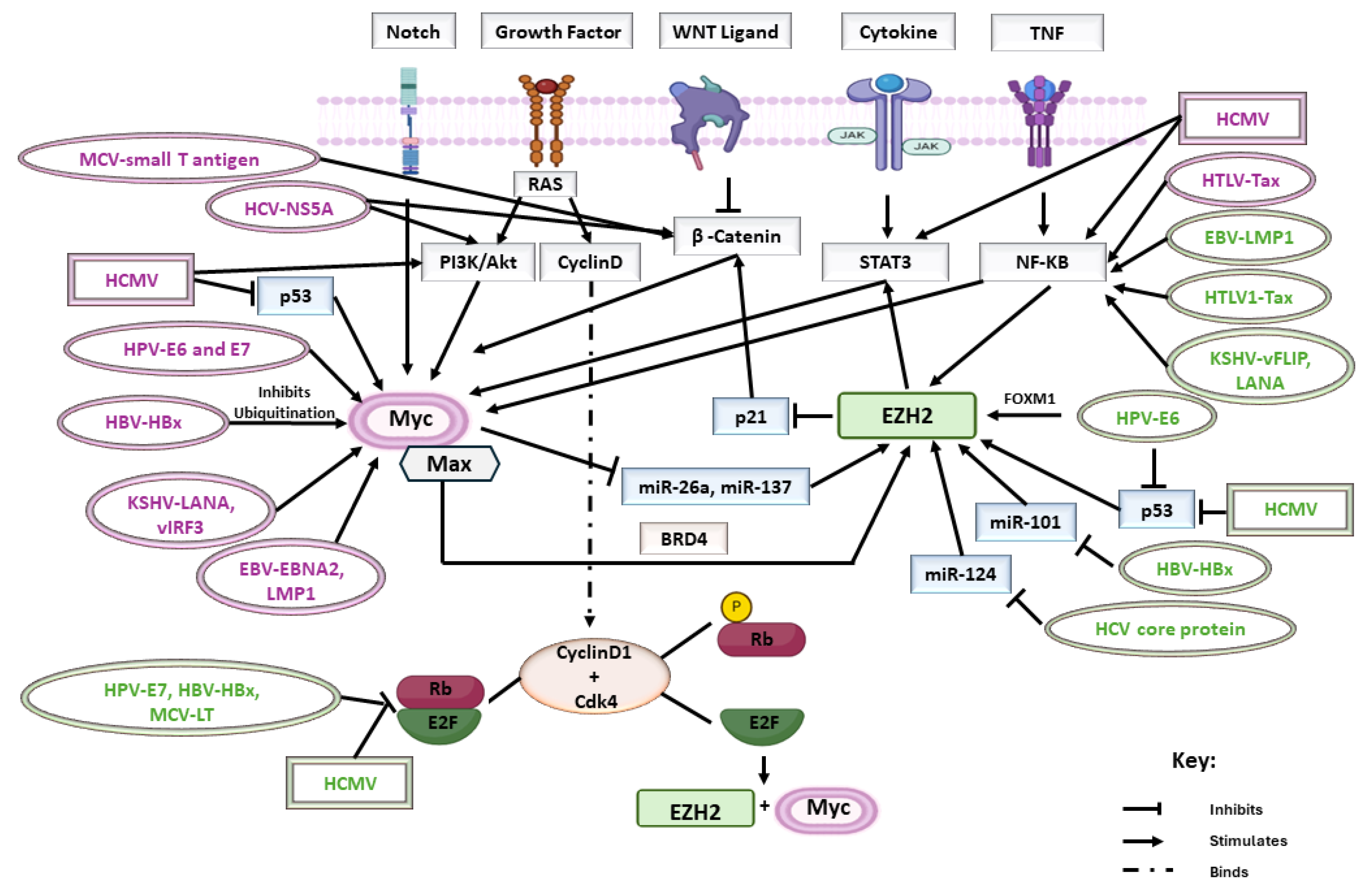
| HPV | HPV E6 | -Increased the levels of the transcription factor FOXM1 and promoted EZH2 and H3K27me3 expression -E6-mediated loss of p53 led to increased EZH2 expression | HPV E6 and HPV E7 | -E6 as well as E7 interacted with c-Myc, leading to the activation of the hTERT promoter -Activation of the hTERT promoter by E6 necessitates Myc binding sites (E boxes) on the hTERT promoter -E7 formed a complex with the myc-interacting zinc-finger protein-1 (Miz-1) |
| HPV E7 | -Activated E2F1 by binding to Rb, thus promoting EZH2 and H3K27me3 expression | |||
| HTLV | Tax | -MAPK- and NFκB-dependent mechanism led to elevated EZH2 protein levels | Tax | -Induced the transcription of Myc by activating NF-κB |
| KSHV | vFLIP and LANA | -Activated the NF-κB pathway leading to increased expression of EZH2 | LANA and vIRF3 | -Stabilized Myc at the post-translational level |
| HBV | HBx | -Elevated EZH2 expression by diminishing miR-101 -Inhibited Rb, resulting in E2F1-mediated transcription of the EZH2 gene | HBx | -Inhibited the ubiquitination of Myc |
| HCV | HCV core protein | -Influenced H3K27me3 levels through a miR-124/EZH2 pathway | HCV non-structural protein NS5A | -Activated Akt -Stabilization of the transcription factor β-catenin, thus activating the c-Myc promoter and increasing c-Myc transcription |
| EBV | LMP1 | -NF-κB activation stimulated EZH2 gene expression | EBNA2 and LMP1 | -EBNA2 induced chromatin loops that connect two enhancers upstream of the Myc transcriptional starting site -LMP1 activated NF-κB; NF-κB is a positive regulator of Myc expression |
| MCPyV | MCV LT | -Inhibition of Rb triggered the activation of E2F transcription factor and EZH2 expression | small T antigen | -Disrupted WNT signaling by elevating the primary key regulator of the β-catenin pathway, thus stimulating Myc synthesis and stabilization |
| HCMV | IE1, IE2, pUL97 | -Increased phosphorylated-Rb (pRb) through the pRB-E2F pathway described to regulate EZH2 expression | IE1, IE2 | -Low p53 levels -Increased Myc, Fos, and Jun expression by IE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Baba, R.; Herbein, G. EZH2-Myc Hallmark in Oncovirus/Cytomegalovirus Infections and Cytomegalovirus’ Resemblance to Oncoviruses. Cells 2024, 13, 541. https://doi.org/10.3390/cells13060541
El Baba R, Herbein G. EZH2-Myc Hallmark in Oncovirus/Cytomegalovirus Infections and Cytomegalovirus’ Resemblance to Oncoviruses. Cells. 2024; 13(6):541. https://doi.org/10.3390/cells13060541
Chicago/Turabian StyleEl Baba, Ranim, and Georges Herbein. 2024. "EZH2-Myc Hallmark in Oncovirus/Cytomegalovirus Infections and Cytomegalovirus’ Resemblance to Oncoviruses" Cells 13, no. 6: 541. https://doi.org/10.3390/cells13060541