
Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease

 , ,
, ,  and
and
Abstract
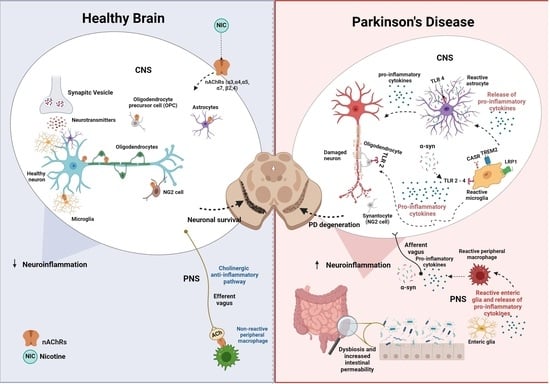
:
{kind=link}
{kind=link}
{kind=link}
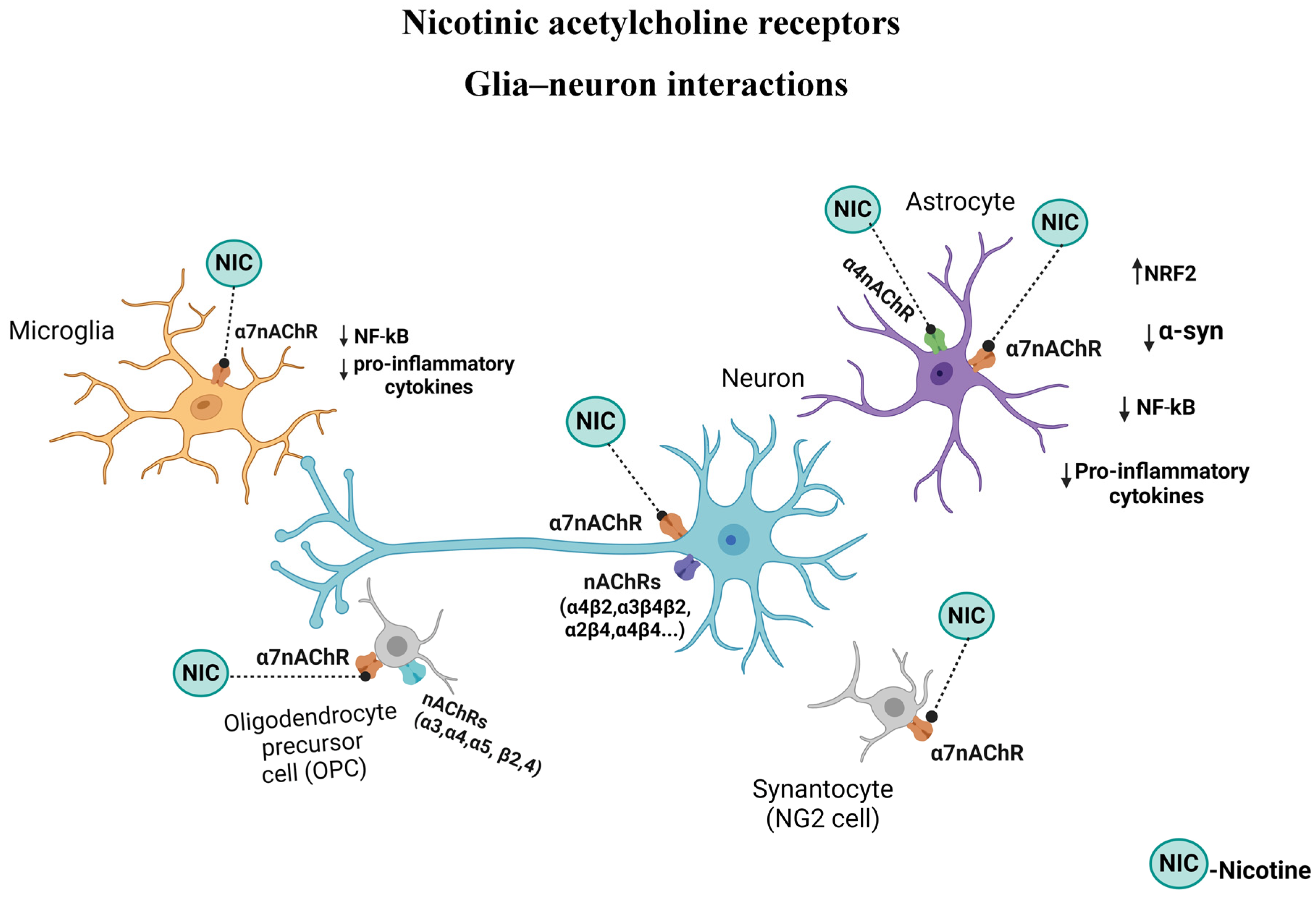{kind=link}
1. Introduction
2. PD Pathophysiology and Current Treatments
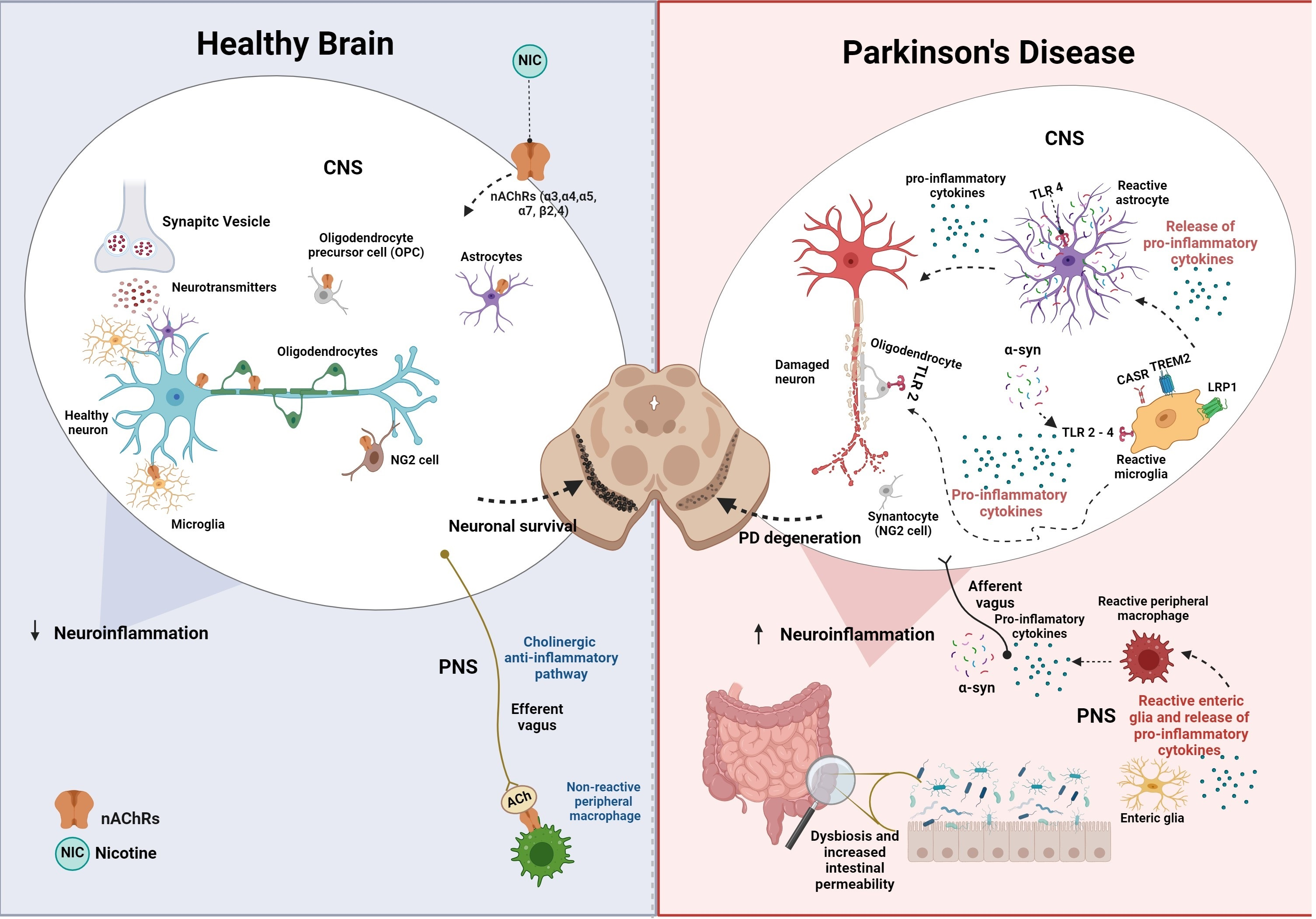
3. Glial Cells
4. Microglia
5. Microglia Synucleinopathies
6. Astroglia
7. Oligodendrocytes
8. Synantocytes (NG2 Cells)
9. Nicotine
10. nAChRs
11. Nicotine for PD
12. Mode of Nicotine Administration as a Critical Factor in PD
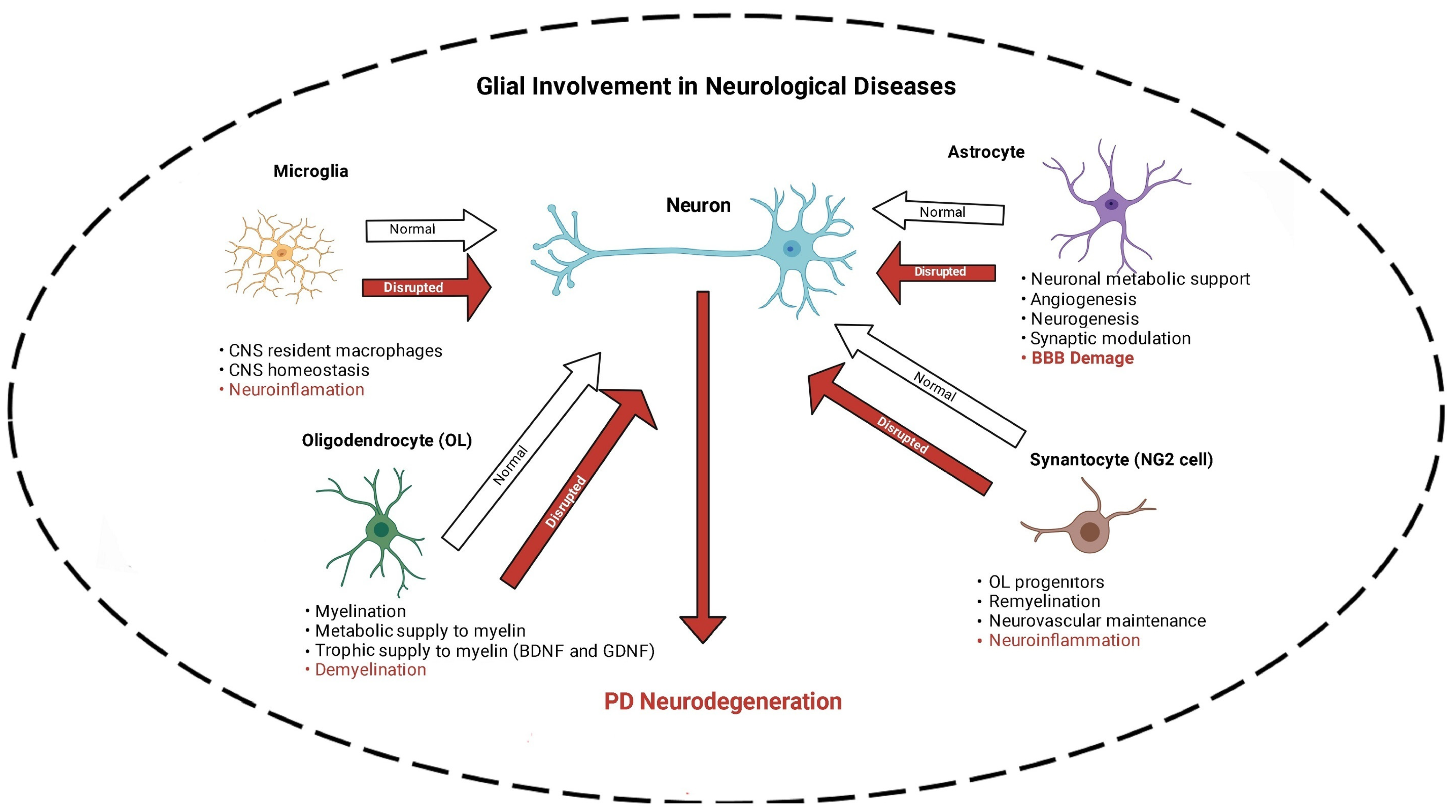
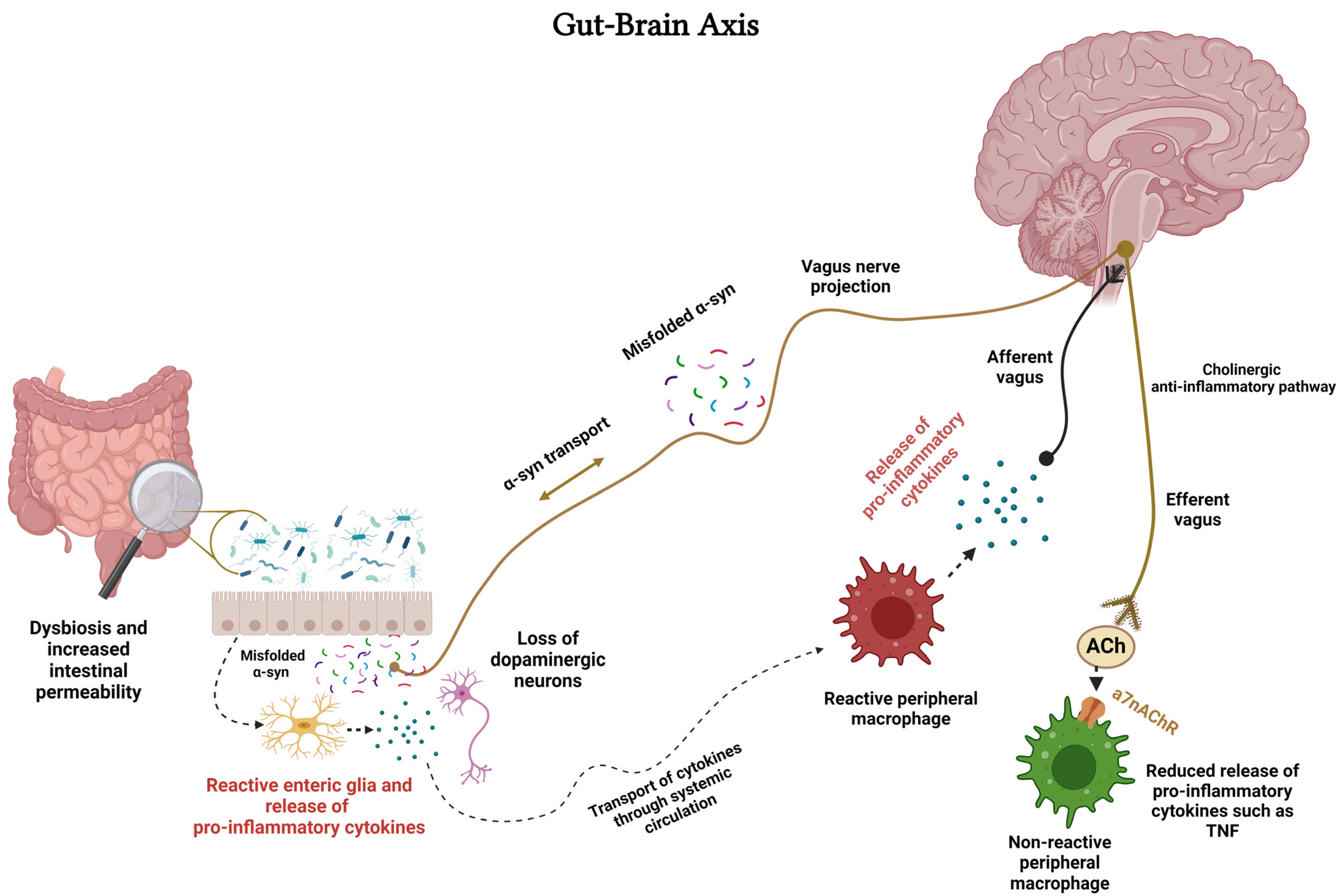
13. nAChR–Microglia and Gut–Brain Axis
14. nAChR–Astroglia
15. nAChR–Oligodendrocyte (OL)
16. nAChRs–NG2 Cells
17. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Parkinson Disease. Available online: https://www.who.int/news-room/fact-sheets/detail/parkinson-disease (accessed on 26 August 2022).
- Snijders, A.H.; Takakusaki, K.; Debu, B.; Lozano, A.M.; Krishna, V.; Fasano, A.; Aziz, T.Z.; Papa, S.M.; Factor, S.A.; Hallett, M. Physiology of Freezing of Gait. Ann. Neurol. 2016, 80, 644–659. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Aschner, M. Novel Pharmacotherapies in Parkinson’s Disease. Neurotox. Res. 2021, 39, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Vallés, A.S.; Barrantes, F.J. Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases. Cells 2023, 12, 2051. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The Genetic Background of Parkinson’s Disease: Current Progress and Future Prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The Genetics of Parkinson Disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Cherian, A.; Divya, K.P.; Vijayaraghavan, A. Parkinson’s Disease—Genetic Cause. Curr. Opin. Neurol. 2023, 36, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Reichmann, H. Considerations on the Role of Environmental Toxins in Idiopathic Parkinson’s Disease Pathophysiology. Transl. Neurodegener. 2014, 3, 10. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Mannervik, B. A Preclinical Model for Parkinson’s Disease Based on Transcriptional Gene Activation via KEAP1/NRF2 to Develop New Antioxidant Therapies. Antioxidants 2023, 12, 673. [Google Scholar] [CrossRef]
- Voon, S.M.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Voon, K.G.L.; Yap, Y.J.; Koh, R.Y. The Mechanism of Action of Salsolinol in Brain: Implications in Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2021, 19, 725–740. [Google Scholar] [CrossRef]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.H.T.P.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini. Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial Dysfunction in Parkinson’s Disease—A Key Disease Hallmark with Therapeutic Potential. Mol. Neurodegener. 2023, 18, 83. [Google Scholar] [CrossRef]
- Lizama, B.N.; Chu, C.T. Neuronal Autophagy and Mitophagy in Parkinson’s Disease. Mol. Asp. Med. 2021, 82, 100972. [Google Scholar] [CrossRef] [PubMed]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-First versus Body-First Parkinson’s Disease: A Multimodal Imaging Case-Control Study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, H.; Ilyas, I.; Mahmood, A.; Hou, L. Microglia and Astrocytes Dysfunction and Key Neuroinflammation-Based Biomarkers in Parkinson’s Disease. Brain Sci. 2023, 13, 634. [Google Scholar] [CrossRef] [PubMed]
- Górska, A.; Markiewicz-Gospodarek, A.; Markiewicz, R.; Chilimoniuk, Z.; Borowski, B.; Trubalski, M.; Czarnek, K. Distribution of Iron, Copper, Zinc and Cadmium in Glia, Their Influence on Glial Cells and Relationship with Neurodegenerative Diseases. Brain Sci. 2023, 13, 911. [Google Scholar] [CrossRef] [PubMed]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Pajarillo, E.; Nyarko-Danquah, I.; Aschner, M.; Lee, E. Role of Astrocytes in Parkinson’s Disease Associated with Genetic Mutations and Neurotoxicants. Cells 2023, 12, 622. [Google Scholar] [CrossRef]
- Piovesana, R.; Reid, A.J.; Tata, A.M. Emerging Roles of Cholinergic Receptors in Schwann Cell Development and Plasticity. Biomedicines 2022, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Hustad, E.; Aasly, J.O. Clinical and Imaging Markers of Prodromal Parkinson’s Disease. Front. Neurol. 2020, 11, 395. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Prajjwal, P.; Flores Sanga, H.S.; Acharya, K.; Tango, T.; John, J.; Rodriguez, R.S.C.; Dheyaa Marsool Marsool, M.; Sulaimanov, M.; Ahmed, A.; Hussin, O.A. Parkinson’s Disease Updates: Addressing the Pathophysiology, Risk Factors, Genetics, Diagnosis, along with the Medical and Surgical Treatment. Ann. Med. Surg. 2023, 85, 4887–4902. [Google Scholar] [CrossRef]
- Quik, M.; Boyd, J.T.; Bordia, T.; Perez, X. Potential Therapeutic Application for Nicotinic Receptor Drugs in Movement Disorders. Nicotine Tob. Res. 2019, 21, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Carroll, V.; Rossiter, R.; Blanchard, D. Non-Motor Symptoms of Parkinson’s Disease. Aust. J. Gen. Pract. 2021, 50, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Onofrj, M.; Russo, M.; Carrarini, C.; Delli Pizzi, S.; Thomas, A.; Bonanni, L.; Espay, A.J.; Sensi, S.L. Functional Neurological Disorder and Somatic Symptom Disorder in Parkinson’s Disease. J. Neurol. Sci. 2022, 433, 120017. [Google Scholar] [CrossRef]
- di Biase, L.; Pecoraro, P.M.; Carbone, S.P.; Caminiti, M.L.; Di Lazzaro, V. Levodopa-Induced Dyskinesias in Parkinson’s Disease: An Overview on Pathophysiology, Clinical Manifestations, Therapy Management Strategies and Future Directions. J. Clin. Med. 2023, 12, 4427. [Google Scholar] [CrossRef] [PubMed]
- Bove, F.; Angeloni, B.; Sanginario, P.; Rossini, P.M.; Calabresi, P.; Di Iorio, R. Neuroplasticity in Levodopa-Induced Dyskinesias: An Overview on Pathophysiology and Therapeutic Targets. Prog. Neurobiol. 2024, 232, 102548. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Pal, G. Initial Management of Parkinson’s Disease. BMJ 2014, 349, g6258. [Google Scholar] [CrossRef] [PubMed]
- Tarakad, A.; Jankovic, J. Diagnosis and Management of Parkinson’s Disease. Semin. Neurol. 2017, 37, 118–126. [Google Scholar] [CrossRef]
- Cardoso, F. Tratamento Da Doença de Parkinson. Arq. Neuropsiquiatr. 1995, 53, 1–10. [Google Scholar] [CrossRef]
- Malek, N. Deep Brain Stimulation in Parkinson’s Disease. Neurol. India 2019, 67, 968. [Google Scholar] [CrossRef]
- Bye, L.J.; Finol-Urdaneta, R.K.; Tae, H.-S.; Adams, D.J. Nicotinic Acetylcholine Receptors: Key Targets for Attenuating Neurodegenerative Diseases. Int. J. Biochem. Cell Biol. 2023, 157, 106387. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Mendoza, C.; Iarkov, A. Nicotinic Acetylcholine Receptors and Learning and Memory Deficits in Neuroinflammatory Diseases. Front. Neurosci. 2023, 17, 1179611. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Huang, S. Comparative Insight into Microglia/Macrophages-Associated Pathways in Glioblastoma and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 25, 16. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The Astrocyte Biochemistry. Semin. Cell Dev. Biol. 2019, 95, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Bonvento, G.; Bolaños, J.P. Astrocyte-Neuron Metabolic Cooperation Shapes Brain Activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.D.; Copperi, F.; Diano, S. Microglia in Central Control of Metabolism. Physiology 2024, 39, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. [Google Scholar] [CrossRef]
- Fernandes, V.M.; Auld, V.; Klämbt, C. Glia as Functional Barriers and Signaling Intermediaries. Cold Spring Harb. Perspect. Biol. 2024, 16, a041423. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Novikov, N.I.; Brazhnik, E.S.; Kitchigina, V.F. Pathological Correlates of Cognitive Decline in Parkinson’s Disease: From Molecules to Neural Networks. Biochemistry 2023, 88, 1890–1904. [Google Scholar] [CrossRef]
- Fiacco, T.A.; Mccarthy, K.D.; Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The Tripartite Glutamatergic Synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef]
- Rasia-Filho, A.A.; Calcagnotto, M.E.; von Bohlen und Halbach, O. (Eds.) Dendritic Spines; Springer International Publishing: Cham, Switzerland, 2023; Volume 34, ISBN 978-3-031-36158-6. [Google Scholar]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide Detoxification by Brain Cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2014, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Reed, M.M.; Blazer-Yost, B. Channels and Transporters in Astrocyte Volume Regulation in Health and Disease. Cell Physiol. Biochem. 2022, 56, 12–30. [Google Scholar] [CrossRef]
- Ebling, F.J.P.; Lewis, J.E. Tanycytes and Hypothalamic Control of Energy Metabolism. Glia 2018, 66, 1176–1184. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Huang, N.; Xie, Y.; LiCausi, F.; Li, S.; Li, Y.; Sheng, Z.H. Oligodendrocytes Enhance Axonal Energy Metabolism by Deacetylation of Mitochondrial Proteins through Transcellular Delivery of SIRT2. Neuron 2021, 109, 3456–3472.e8. [Google Scholar] [CrossRef]
- Clayton, R.W.; Lovell-Badge, R.; Galichet, C. The Properties and Functions of Glial Cell Types of the Hypothalamic Median Eminence. Front. Endocrinol. 2022, 13, 953995. [Google Scholar] [CrossRef] [PubMed]
- Kofler, J.; Wiley, C.A. Microglia: Key Innate Immune Cells of the Brain. Toxicol. Pathol 2011, 39, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Holtzman, D.M. Emerging Roles of Innate and Adaptive Immunity in Alzheimer’s Disease. Immunity 2022, 55, 2236–2254. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like Receptors 2 and 4 Differentially Regulate the Self-Renewal and Differentiation of Spinal Cord Neural Precursor Cells. Stem. Cell Res. Ther. 2022, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Wies Mancini, V.S.B.; Mattera, V.S.; Pasquini, J.M.; Pasquini, L.A.; Correale, J.D. Microglia-derived Extracellular Vesicles in Homeostasis and Demyelination/Remyelination Processes. J. Neurochem. 2024, 168, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Alzarea, S. Glial Mechanisms Underlying Major Depressive Disorder: Potential Therapeutic Opportunities. Progress Mol. Biol. Transl. Sci. 2019, 167, 159–178. [Google Scholar]
- Scuderi, C.; Verkhratsky, A.; Parpura, V.; Li, B. Neuroglia in Psychiatric Disorders. In Astrocytes in Psychiatric Disorders; Springer: Cham, Switzerland, 2021; pp. 3–19. [Google Scholar]
- Hanslik, K.L.; Marino, K.M.; Ulland, T.K. Modulation of Glial Function in Health, Aging, and Neurodegenerative Disease. Front. Cell Neurosci. 2021, 15, 718324. [Google Scholar] [CrossRef]
- Zhao, G. Shared and Disease-Specific Glial Gene Expression Changes in Neurodegenerative Diseases. Nat. Aging 2023, 3, 246–247. [Google Scholar] [CrossRef]
- Zhu, H.; Guan, A.; Liu, J.; Peng, L.; Zhang, Z.; Wang, S. Noteworthy Perspectives on Microglia in Neuropsychiatric Disorders. J. Neuroinflamm. 2023, 20, 223. [Google Scholar] [CrossRef]
- Costa, T.; Fernandez-Villalba, E.; Izura, V.; Lucas-Ochoa, A.; Menezes-Filho, N.; Santana, R.; de Oliveira, M.; Araújo, F.; Estrada, C.; Silva, V.; et al. Combined 1-Deoxynojirimycin and Ibuprofen Treatment Decreases Microglial Activation, Phagocytosis and Dopaminergic Degeneration in MPTP-Treated Mice. J. Neuroimmune Pharmacol. 2021, 16, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, Z.; Lu, G.; Chan, W.; Zhang, Y.; Wong, G.C. Microglia Activation, Classification and Microglia-Mediated Neuroinflammatory Modulators in Subarachnoid Hemorrhage. Neural. Regen. Res. 2022, 17, 1404. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Sriram, K. Molecular Mechanisms Underlying Neuroinflammation Elicited by Occupational Injuries and Toxicants. Int. J. Mol. Sci. 2023, 24, 2272. [Google Scholar] [CrossRef] [PubMed]
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The Role of Microglia in the Development of Neurodegeneration. Neurol. Sci. 2020, 41, 3609–3615. [Google Scholar] [CrossRef]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The Dual Face of Microglia (M1/M2) as a Potential Target in the Protective Effect of Nutraceuticals against Neurodegenerative Diseases. Front. Aging 2023, 4, 1231706. [Google Scholar] [CrossRef]
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia Activation in Central Nervous System Disorders: A Review of Recent Mechanistic Investigations and Development Efforts. Front. Neurol. 2023, 14, 1103416. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain: Figure 1. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Nimmerjahn, A. Two-Photon Imaging of Microglia in the Mouse Cortex In Vivo. Cold Spring Harb. Protoc. 2012, 2012, pdb.prot069294. [Google Scholar] [CrossRef] [PubMed]
- Leyh, J.; Paeschke, S.; Mages, B.; Michalski, D.; Nowicki, M.; Bechmann, I.; Winter, K. Classification of Microglial Morphological Phenotypes Using Machine Learning. Front. Cell Neurosci. 2021, 15, 701673. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Taylor, S.E.; Cao, T.; Harrison, J.L.; Lifshitz, J. Rod Microglia: Elongation, Alignment, and Coupling to Form Trains across the Somatosensory Cortex after Experimental Diffuse Brain Injury. J. Neuroinflamm. 2012, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod Microglia: A Morphological Definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-Y.; Guo, Y.; Seki, S.M.; Rosen, A.M.; Johanson, D.M.; Mandell, J.W.; Lucchinetti, C.F.; Gaultier, A. LRP1 Expression in Microglia Is Protective during CNS Autoimmunity. Acta Neuropathol. Commun. 2016, 4, 68. [Google Scholar] [CrossRef]
- de Araújo, F.M.; Cuenca-Bermejo, L.; Fernández-Villalba, E.; Costa, S.L.; Silva, V.D.A.; Herrero, M.T. Role of Microgliosis and NLRP3 Inflammasome in Parkinson’s Disease Pathogenesis and Therapy. Cell Mol. Neurobiol. 2022, 42, 1283–1300. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Wu, F.; Zhan, R.-Y.; Zhou, H.-J. Inflammatory Role of Microglia in Brain Injury Caused by Subarachnoid Hemorrhage. Front. Cell Neurosci. 2022, 16, 956185. [Google Scholar] [CrossRef]
- Fracassi, A.; Marcatti, M.; Tumurbaatar, B.; Woltjer, R.; Moreno, S.; Taglialatela, G. TREM2-induced Activation of Microglia Contributes to Synaptic Integrity in Cognitively Intact Aged Individuals with Alzheimer’s Neuropathology. Brain Pathol. 2023, 33, e13108. [Google Scholar] [CrossRef]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Microglia as Therapeutic Target in Central Nervous System Disorders. J. Pharmacol. Sci. 2020, 144, 102–118. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-Astrocyte Omnidirectional Signaling in Neurological Health and Disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef]
- Hoogland, I.C.M.; Yik, J.; Westhoff, D.; Engelen-Lee, J.-Y.; Valls Seron, M.; Man, W.K.; Houben-Weerts, J.H.P.M.; Tanck, M.W.T.; van Westerloo, D.J.; van der Poll, T.; et al. Microglial Cell Response in α7 Nicotinic Acetylcholine Receptor-Deficient Mice after Systemic Infection with Escherichia Coli. J. Neuroinflamm. 2022, 19, 94. [Google Scholar] [CrossRef] [PubMed]
- Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int. J. Mol. Sci. 2023, 24, 2477. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Meng, H. The Involvement of α-Synucleinopathy in the Disruption of Microglial Homeostasis Contributes to the Pathogenesis of Parkinson’s Disease. Cell Commun. Signal 2024, 22, 31. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.V.; Landis, H.E.; Getachew, B.; Diogenes Amaral Silva, V.; Ribeiro, P.R.; Aschner, M.; Tizabi, Y. Iron Toxicity, Ferroptosis and Microbiota in Parkinson’s Disease: Implications for Novel Targets. Adv. Neurotoxicol. 2024, 11, 2468–7480. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, W.; Sun, Y.; Wu, M. New Insight on Microglia Activation in Neurodegenerative Diseases and Therapeutics. Front. Neurosci. 2023, 17, 1308345. [Google Scholar] [CrossRef] [PubMed]
- Saramowicz, K.; Siwecka, N.; Galita, G.; Kucharska-Lusina, A.; Rozpędek-Kamińska, W.; Majsterek, I. Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 25, 360. [Google Scholar] [CrossRef]
- Sharma, A.; Lee, S.; Kim, H.; Yoon, H.; Ha, S.; Kang, S.U. Molecular Crosstalk Between Circadian Rhythmicity and the Development of Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 844. [Google Scholar] [CrossRef]
- Guzmán-Ruiz, M.A.; Guerrero-Vargas, N.N.; Lagunes-Cruz, A.; González-González, S.; García-Aviles, J.E.; Hurtado-Alvarado, G.; Mendez-Hernández, R.; Chavarría-Krauser, A.; Morin, J.; Arriaga-Avila, V.; et al. Circadian Modulation of Microglial Physiological Processes and Immune Responses. Glia 2023, 71, 155–167. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like Receptor 4 Is Required for α-synuclein Dependent Activation of Microglia and Astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of Astroglial Toll-like Receptors (TLRs) in Central Nervous System Infections, Injury and Neurodegenerative Diseases. Brain Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef]
- Parra-Rivas, L.A.; Madhivanan, K.; Aulston, B.D.; Wang, L.; Prakashchand, D.D.; Boyer, N.P.; Saia-Cereda, V.M.; Branes-Guerrero, K.; Pizzo, D.P.; Bagchi, P.; et al. Serine-129 Phosphorylation of α-Synuclein Is an Activity-Dependent Trigger for Physiologic Protein-Protein Interactions and Synaptic Function. Neuron 2023, 111, 4006–4023.e10. [Google Scholar] [CrossRef]
- Harvey, J.; Pishva, E.; Chouliaras, L.; Lunnon, K. Elucidating Distinct Molecular Signatures of Lewy Body Dementias. Neurobiol. Dis. 2023, 188, 106337. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Yang, B.; Chen, Q.; Zhang, J.; Hu, J.; Fan, Y. Activation of A7 Nicotinic Acetylcholine Receptor Protects Against 1-Methyl-4-Phenylpyridinium-Induced Astroglial Apoptosis. Front. Cell Neurosci. 2019, 13, 507. [Google Scholar] [CrossRef]
- Aryal, S.P.; Fu, X.; Sandin, J.N.; Neupane, K.R.; Lakes, J.E.; Grady, M.E.; Richards, C.I. Nicotine Induces Morphological and Functional Changes in Astrocytes via Nicotinic Receptor Activity. Glia 2021, 69, 2037–2053. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Schneider, A.M.; Mulloy, S.M.; Lee, A.M. Expression Pattern of Nicotinic Acetylcholine Receptor Subunit Transcripts in Neurons and Astrocytes in the Ventral Tegmental Area and Locus Coeruleus. Eur. J. Neurosci. 2023. early view. [Google Scholar] [CrossRef]
- Ma, W.; Si, T.; Wang, Z.; Wen, P.; Zhu, Z.; Liu, Q.; Wang, J.; Xu, F.; Li, Q. Astrocytic α4-Containing NAChR Signaling in the Hippocampus Governs the Formation of Temporal Association Memory. Cell Rep. 2023, 42, 112674. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W. Strategies for Metabolic Exchange between Glial Cells and Neurons. Respir. Physiol. 2001, 129, 71–81. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-Synuclein Immunoreactive Astrocytes in the Forebrain Parallels Stages of Intraneuronal Pathology in Sporadic Parkinson’s Disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.T.; Ureshino, R.P.; et al. Overexpression of α-Synuclein in an Astrocyte Cell Line Promotes Autophagy Inhibition and Apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef]
- Huenchuguala, S.; Munõz, P.; Zavala, P.; Villa, M.; Cuevas, C.; Ahumada, U.; Graumann, R.; Nore, B.F.; Couve, E.; Mannervik, B.; et al. Glutathione Transferase Mu 2 Protects Glioblastoma Cells against Aminochrome Toxicity by Preventing Autophagy and Lysosome Dysfunction. Autophagy 2014, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Huenchuguala, S.; Muñoz, P.; Graumann, R.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Protects Astrocytes from Aminochrome-Induced Toxicity. Neurotoxicology 2016, 55, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione Transferase-M2-2 Secreted from Glioblastoma Cell Protects SH-SY5Y Cells from Aminochrome Neurotoxicity. Neurotox. Res. 2015, 27, 217–228. [Google Scholar] [CrossRef]
- Valdes, R.; Armijo, A.; Muñoz, P.; Hultenby, K.; Hagg, A.; Inzunza, J.; Nalvarte, I.; Varshney, M.; Mannervik, B.; Segura-Aguilar, J. Cellular Trafficking of Glutathione Transferase M2-2 Between U373MG and SHSY-S7 Cells Is Mediated by Exosomes. Neurotox. Res. 2021, 39, 182–190. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Muñoz, P.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Mannervik, B. Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase. Antioxidants 2022, 11, 296. [Google Scholar] [CrossRef]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Xu, Z.-Q.; Zhang, W.-J.; Su, D.-F.; Zhang, G.-Q.; Miao, C.-Y. Cellular Responses and Functions of α7 Nicotinic Acetylcholine Receptor Activation in the Brain: A Narrative Review. Ann. Transl. Med. 2021, 9, 509. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.-P.; Kothary, R. Oligodendrocytes in a Nutshell. Front. Cell Neurosci. 2015, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; He, C.; Zhang, S.; Zhao, Y.; Zhu, M.; Tang, X.; Li, Q.; Xu, L.; Yang, Y. Progress in Methods for Evaluating Schwann Cell Myelination and Axonal Growth in Peripheral Nerve Regeneration via Scaffolds. Front. Bioeng. Biotechnol. 2023, 11, 1308761. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.; Tadi, P. Histology, Schwann Cells; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A Single-Cell Atlas of the Human Substantia Nigra Reveals Cell-Specific Pathways Associated with Neurological Disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef]
- Bae, E.-J.; Pérez-Acuña, D.; Rhee, K.H.; Lee, S.-J. Changes in Oligodendroglial Subpopulations in Parkinson’s Disease. Mol. Brain 2023, 16, 65. [Google Scholar] [CrossRef]
- Menichella, D.M.; Majdan, M.; Awatramani, R.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Genetic and Physiological Evidence That Oligodendrocyte Gap Junctions Contribute to Spatial Buffering of Potassium Released during Neuronal Activity. J. Neurosci. 2006, 26, 10984. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J. Neuronal Activity and Remyelination: New Insights into the Molecular Mechanisms and Therapeutic Advancements. Front. Cell Dev. Biol. 2023, 11, 1221890. [Google Scholar] [CrossRef]
- Bsibsi, M.; Nomden, A.; van Noort, J.M.; Baron, W. Toll-like Receptors 2 and 3 Agonists Differentially Affect Oligodendrocyte Survival, Differentiation, and Myelin Membrane Formation. J. Neurosci. Res. 2012, 90, 388–398. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like Receptors in the Pathogenesis of Neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Hill, R.A.; Nishiyama, A. NG2 Cells (Polydendrocytes): Listeners to the Neural Network with Diverse Properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef]
- Kirdajova, D.; Anderova, M. NG2 Cells and Their Neurogenic Potential. Curr. Opin. Pharmacol. 2020, 50, 53–60. [Google Scholar] [CrossRef]
- Xu, J.-P.; Zhao, J.; Li, S. Roles of NG2 Glial Cells in Diseases of the Central Nervous System. Neurosci. Bull. 2011, 27, 413–421. [Google Scholar] [CrossRef]
- Dimou, L.; Gallo, V. NG2-glia and Their Functions in the Central Nervous System. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a Common Denominator for Neuroinflammation, Blood–Brain Barrier Alteration, and Oligodendrocyte Precursor Response in EAE, Plays a Role in Dendritic Cell Activation. Acta Neuropathol. 2016, 132, 23–42. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Q.; Yang, Q.; Gu, H.; Yin, Y.; Li, Y.; Hou, J.; Chen, R.; Sun, Q.; Sun, Y.; et al. NG2 Glia Regulate Brain Innate Immunity via TGF-Β2/TGFBR2 Axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef]
- Hu, X.; Geng, P.; Zhao, X.; Wang, Q.; Liu, C.; Guo, C.; Dong, W.; Jin, X. The NG2-Glia Is a Potential Target to Maintain the Integrity of Neurovascular Unit after Acute Ischemic Stroke. Neurobiol. Dis. 2023, 180, 106076. [Google Scholar] [CrossRef]
- Vélez-Fort, M.; Audinat, E.; Angulo, M.C. Functional α7-containing Nicotinic Receptors of NG2-expressing Cells in the Hippocampus. Glia 2009, 57, 1104–1114. [Google Scholar] [CrossRef]
- Timmermann, A.; Tascio, D.; Jabs, R.; Boehlen, A.; Domingos, C.; Skubal, M.; Huang, W.; Kirchhoff, F.; Henneberger, C.; Bilkei-Gorzo, A.; et al. Dysfunction of NG2 Glial Cells Affects Neuronal Plasticity and Behavior. Glia 2023, 71, 1481–1501. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gil, G.F.; Reitsma, M.B.; Ahmad, N.S.; Anderson, J.A.; Bisignano, C.; Carr, S.; Feldman, R.; Hay, S.I.; He, J.; et al. Health Effects Associated with Smoking: A Burden of Proof Study. Nat. Med. 2022, 28, 2045–2055. [Google Scholar] [CrossRef]
- Dani, J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int. Rev. Neurobiol. 2015, 124, 3. [Google Scholar] [CrossRef]
- Papke, R.L.; Lindstrom, J.M. Nicotinic Acetylcholine Receptors: Conventional and Unconventional Ligands and Signaling. Neuropharmacology 2020, 168, 108021. [Google Scholar] [CrossRef]
- Cecchini, M.; Changeux, J.P. Nicotinic Receptors: From Protein Allostery to Computational Neuropharmacology. Mol. Asp. Med. 2022, 84, 101044. [Google Scholar] [CrossRef]
- Nara, S.; Yamaguti, Y.; Tsuda, I. Review: Nicotinic Acetylcholine Receptors to Regulate Important Brain Activity—What Occurs at the Molecular Level? Cogn. Neurodyn. 2023, 19, 1–16. [Google Scholar] [CrossRef]
- Wonnacott, S. Nicotinic Receptors. In Encyclopedia of Molecular Pharmacology; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–7. [Google Scholar]
- Tizabi, Y.; Getachew, B.; Tsytsarev, V.; Csoka, A.B.; Copeland, R.L.; Heinbockel, T. Central Nicotinic and Muscarinic Receptors in Health and Disease. In Acetylcholine—Recent Advances and New Perspectives; IntechOpen: London, UK, 2023. [Google Scholar]
- Kasheverov, I.; Kudryavtsev, D.; Shelukhina, I.; Nikolaev, G.; Utkin, Y.; Tsetlin, V. Marine Origin Ligands of Nicotinic Receptors: Low Molecular Compounds, Peptides and Proteins for Fundamental Research and Practical Applications. Biomolecules 2022, 12, 189. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef]
- Mugayar, A.A.; da Silva Guimarães, G.; de Oliveira, P.H.T.; Miranda, R.L.; dos Santos, A.A. Apoptosis in the Neuroprotective Effect of α7 Nicotinic Receptor in Neurodegenerative Models. J. Neurosci. Res. 2023, 101, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Keever, K.R.; Yakubenko, V.P.; Hoover, D.B. Neuroimmune Nexus in the Pathophysiology and Therapy of Inflammatory Disorders: Role of α7 Nicotinic Acetylcholine Receptors. Pharmacol. Res. 2023, 191, 106758. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, P.; Kabbani, N. Ionotropic and Metabotropic Responses by Alpha 7 Nicotinic Acetylcholine Receptors. Pharmacol. Res. 2023, 197, 106975. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic Acetylcholine Receptors: Therapeutic Targets for Novel Ligands to Treat Pain and Inflammation. Pharmacol. Res. 2023, 190, 106715. [Google Scholar] [CrossRef]
- Salehi, Z.; Motlagh Ghoochani, B.F.N.; Hasani Nourian, Y.; Jamalkandi, S.A.; Ghanei, M. The Controversial Effect of Smoking and Nicotine in SARS-CoV-2 Infection. Allergy Asthma. Clin. Immunol. 2023, 19, 49. [Google Scholar] [CrossRef]
- Shelukhina, I.; Siniavin, A.; Kasheverov, I.; Ojomoko, L.; Tsetlin, V.; Utkin, Y. α7- and α9-Containing Nicotinic Acetylcholine Receptors in the Functioning of Immune System and in Pain. Int. J. Mol. Sci. 2023, 24, 6524. [Google Scholar] [CrossRef]
- Wills, L.; Ables, J.L.; Braunscheidel, K.M.; Caligiuri, S.P.B.; Elayouby, K.S.; Fillinger, C.; Ishikawa, M.; Moen, J.K.; Kenny, P.J. Neurobiological Mechanisms of Nicotine Reward and Aversion. Pharmacol. Rev. 2022, 74, 271–310. [Google Scholar] [CrossRef]
- Kim, K.; Picciotto, M.R. Nicotine Addiction: More than Just Dopamine. Curr. Opin. Neurobiol. 2023, 83, 102797. [Google Scholar] [CrossRef]
- Sansone, L.; Milani, F.; Fabrizi, R.; Belli, M.; Cristina, M.; Zagà, V.; de Iure, A.; Cicconi, L.; Bonassi, S.; Russo, P. Nicotine: From Discovery to Biological Effects. Int. J. Mol. Sci. 2023, 24, 14570. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Csoka, A.B.; Manaye, K.F.; Copeland, R.L. Novel Targets for Parkinsonism-Depression Comorbidity. Prog. Mol. Biol. Transl. Sci. 2019, 167, 1–24. [Google Scholar] [CrossRef]
- Liu, C. Targeting the Cholinergic System in Parkinson’s Disease. Acta Pharmacol. Sin. 2020, 41, 453–463. [Google Scholar] [CrossRef]
- Gandelman, J.A.; Newhouse, P.; Taylor, W.D. Nicotine and Networks: Potential for Enhancement of Mood and Cognition in Late-Life Depression. Neurosci. Biobehav. Rev. 2018, 84, 289–298. [Google Scholar] [CrossRef]
- Conti, A.A.; Tolomeo, S.; Steele, J.D.; Baldacchino, A.M. Severity of Negative Mood and Anxiety Symptoms Occurring during Acute Abstinence from Tobacco: A Systematic Review and Meta-Analysis. Neurosci. Biobehav. Rev. 2020, 115, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Louis, V.A.; Taylor, C.T.; Waxman, D.; Culver, K.E.; Szechtman, H. Effect of Nicotine on Quinpirole-Induced Checking Behavior in Rats: Implications for Obsessive-Compulsive Disorder. Biol. Psychiatry 2002, 51, 164–171. [Google Scholar] [CrossRef]
- Mitra, S.; Mucha, M.; Khatri, S.N.; Glenon, R.; Schulte, M.K.; Bult-Ito, A. Attenuation of Compulsive-Like Behavior Through Positive Allosteric Modulation of A4β2 Nicotinic Acetylcholine Receptors in Non-Induced Compulsive-Like Mice. Front. Behav. Neurosci. 2017, 10, 244. [Google Scholar] [CrossRef]
- Taylor, M.R.; Carrasco, K.; Carrasco, A.; Basu, A. Tobacco and ADHD: A Role of MAO-Inhibition in Nicotine Dependence and Alleviation of ADHD Symptoms. Front. Neurosci. 2022, 16, 845646. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Russell, L.T.; Johnson, M.; Darmani, N.A. Nicotine Attenuates DOI-Induced Head-Twitch Response in Mice: Implications for Tourette Syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Hayslett, R.L.; Tizabi, Y. Effects of Donepezil, Nicotine and Haloperidol on the Central Serotonergic System in Mice: Implications for Tourette’s Syndrome. Pharmacol. Biochem. Behav. 2005, 81, 879–886. [Google Scholar] [CrossRef]
- Quik, M.; Zhang, D.; McGregor, M.; Bordia, T. Alpha7 Nicotinic Receptors as Therapeutic Targets for Parkinson’s Disease. Biochem. Pharmacol. 2015, 97, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Koukouli, F.; Changeux, J.P. Do Nicotinic Receptors Modulate High-Order Cognitive Processing? Trends Neurosci. 2020, 43, 550–564. [Google Scholar] [CrossRef]
- Nop, O.; Senft Miller, A.; Culver, H.; Makarewicz, J.; Dumas, J.A. Nicotine and Cognition in Cognitively Normal Older Adults. Front. Aging Neurosci. 2021, 13, 640674. [Google Scholar] [CrossRef]
- Hahn, B.; Harvey, A.N.; Concheiro-Guisan, M.; Huestis, M.A.; Ross, T.J.; Stein, E.A. Nicotinic Receptor Modulation of the Default Mode Network. Psychopharmacology 2021, 238, 589–597. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, T.; Zhao, C.; Li, G. The Regulation of Exosome Generation and Function in Physiological and Pathological Processes. Int. J. Mol. Sci. 2023, 25, 255. [Google Scholar] [CrossRef]
- Han, T.; Wang, Q.; Lai, R.; Zhang, D.; Diao, Y.; Yin, Y. Nicotine Induced Neurocognitive Protection and Anti-Inflammation Effect by Activating α4β2 Nicotinic Acetylcholine Receptors in Ischemic Rats. Nicotine Tob. Res. 2020, 22, 919–924. [Google Scholar] [CrossRef]
- Ionov, I.D.; Pushinskaya, I.I.; Gorev, N.P.; Frenkel, D.D.; Severtsev, N.N. Anticataleptic Activity of Nicotine in Rats: Involvement of the Lateral Entorhinal Cortex. Psychopharmacology 2021, 238, 2471–2483. [Google Scholar] [CrossRef]
- Terry, A.V.; Callahan, P.M. α7 Nicotinic Acetylcholine Receptors as Therapeutic Targets in Schizophrenia: Update on Animal and Clinical Studies and Strategies for the Future. Neuropharmacology 2020, 170, 108053. [Google Scholar] [CrossRef]
- Bagdas, D.; Gurun, M.S.; Flood, P.; Papke, R.L.; Damaj, M.I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on α7 NAChRs. Curr. Neuropharmacol. 2018, 16, 415. [Google Scholar] [CrossRef]
- Seoane-Collazo, P.; Diéguez, C.; Nogueiras, R.; Rahmouni, K.; Fernández-Real, J.M.; López, M. Nicotine’ Actions on Energy Balance: Friend or Foe? Pharmacol. Ther. 2021, 219, 107693. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Kenny, P.J. Central and Peripheral Actions of Nicotine That Influence Blood Glucose Homeostasis and the Development of Diabetes. Pharmacol. Res. 2023, 194, 106860. [Google Scholar] [CrossRef]
- Pechlivanidou, M.; Ninou, E.; Karagiorgou, K.; Tsantila, A.; Mantegazza, R.; Francesca, A.; Furlan, R.; Dudeck, L.; Steiner, J.; Tzartos, J.; et al. Autoimmunity to Neuronal Nicotinic Acetylcholine Receptors. Pharmacol. Res. 2023, 192, 106790. [Google Scholar] [CrossRef]
- Arnold, E.C.; Soler-Llavina, G.; Kambara, K.; Bertrand, D. The Importance of Ligand Gated Ion Channels in Sleep and Sleep Disorders. Biochem. Pharmacol. 2023, 212, 115532. [Google Scholar] [CrossRef]
- Lunney, P.C.; Leong, R.W.L. Review Article: Ulcerative Colitis, Smoking and Nicotine Therapy. Aliment. Pharmacol. Ther. 2012, 36, 997–1008. [Google Scholar] [CrossRef]
- Morioka, N.; Hisaoka-Nakashima, K.; Nakata, Y. Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Springer: Cham, Switzerland, 2018; ISBN 9789811084881. [Google Scholar]
- Zhang, W.; Lin, H.; Zou, M.; Yuan, Q.; Huang, Z.; Pan, X.; Zhang, W. Nicotine in Inflammatory Diseases: Anti-Inflammatory and Pro-Inflammatory Effects. Front. Immunol. 2022, 13, 826889. [Google Scholar] [CrossRef]
- Okada, K.; Matsuo, K. Nicotine Exerts a Stronger Immunosuppressive Effect Than Its Structural Analogs and Regulates Experimental Colitis in Rats. Biomedicines 2023, 11, 922. [Google Scholar] [CrossRef]
- Ko, J.; Auyeung, K. Inflammatory Bowel Disease: Etiology, Pathogenesis and Current Therapy. Curr. Pharm. Des. 2014, 20, 1082–1096. [Google Scholar] [CrossRef]
- Gomes, J.P.; Watad, A.; Shoenfeld, Y. Nicotine and Autoimmunity: The Lotus’ Flower in Tobacco. Pharmacol. Res. 2018, 128, 101–109. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Copeland, R.L.; Aschner, M. Nicotine and the Nicotinic Cholinergic System in COVID-19. FEBS J. 2020, 287, 3656–3663. [Google Scholar] [CrossRef]
- Bele, T.; Turk, T.; Križaj, I. Nicotinic Acetylcholine Receptors in Cancer: Limitations and Prospects. Biochim. Biophys. Acta Mol. Basis. Dis. 2024, 1870, 166875. [Google Scholar] [CrossRef]
- Lanciego, J.L.; Luquin, N.; Obeso, J.A. Functional Neuroanatomy of the Basal Ganglia. Cold Spring Harb. Perspect. Med. 2012, 2, a009621. [Google Scholar] [CrossRef]
- Perez, X.A.; Bordia, T.; Quik, M. The Striatal Cholinergic System in L-Dopa-Induced Dyskinesias. J. Neural. Transm. 2018, 125, 1251–1262. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Copeland, R.L.; Moratalla, R.; Patricio, F.; Limón, I.D.; Del Bel, E.; Aschner, M. Novel Pharmacotherapies for L-DOPA-Induced Dyskinesia. In Handbook of Neurotoxicity; Springer International Publishing: Cham, Switzerland, 2021; pp. 1–19. [Google Scholar]
- Copeland, R.L.; Leggett, Y.A.; Kanaan, Y.M.; Taylor, R.E.; Tizabi, Y. Neuroprotective Effects of Nicotine against Salsolinol-Induced Cytotoxicity: Implications for Parkinson’s Disease. Neurotox. Res. 2005, 8, 289–293. [Google Scholar] [CrossRef]
- Getachew, B.; Csoka, A.B.; Bhatti, A.; Copeland, R.L.; Tizabi, Y. Butyrate Protects Against Salsolinol-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurotox. Res. 2020, 38, 596–602. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Aschner, M. Butyrate Protects and Synergizes with Nicotine against Iron- and Manganese-Induced Toxicities in Cell Culture. Neurotox. Res. 2024, 42, 3. [Google Scholar] [CrossRef]
- Getachew, B.; Csoka, A.B.; Aschner, M.; Tizabi, Y. Nicotine Protects against Manganese and Iron-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurochem. Int. 2019, 124, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Cuevas, C.; Villa, M.; Caviedes, P.; Segura-Aguilar, J.; Tizabi, Y. Protective Effects of Nicotine Against Aminochrome-Induced Toxicity in Substantia Nigra Derived Cells: Implications for Parkinson’s Disease. Neurotox. Res. 2012, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, X.; Zhou, X.; Wu, X.; Li, Y.; Yao, J.; Bai, J. Nicotine Suppresses the Neurotoxicity by MPP +/MPTP through Activating α7nAChR/PI3K/Trx-1 and Suppressing ER Stress. Neurotoxicology 2017, 59, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Bi, W.; Zheng, K.; Zhu, E.; Wang, S.; Xiong, Y.; Chang, J.; Jiang, J.; Liu, B.; Lu, Z.; et al. Nicotine Prevents Oxidative Stress-Induced Hippocampal Neuronal Injury Through α7-NAChR/Erk1/2 Signaling Pathway. Front. Mol. Neurosci. 2020, 13, 557647. [Google Scholar] [CrossRef] [PubMed]
- Ruan, S.; Xie, J.; Wang, L.; Guo, L.; Li, Y.; Fan, W.; Ji, R.; Gong, Z.; Xu, Y.; Mao, J.; et al. Nicotine Alleviates MPTP-Induced Nigrostriatal Damage through Modulation of JNK and ERK Signaling Pathways in the Mice Model of Parkinson’s Disease. Front. Pharmacol. 2023, 14, 1088957. [Google Scholar] [CrossRef]
- Fares, M.B.; Alijevic, O.; Johne, S.; Overk, C.; Hashimoto, M.; Kondylis, A.; Adame, A.; Dulize, R.; Peric, D.; Nury, C.; et al. Nicotine-Mediated Effects in Neuronal and Mouse Models of Synucleinopathy. Front. Neurosci. 2023, 17, 1239009. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.I.; Porcu, A.; Romoli, B.; Keisler, M.; Manfredsson, F.P.; Powell, S.B.; Dulcis, D. Nicotine-Mediated Recruitment of GABAergic Neurons to a Dopaminergic Phenotype Attenuates Motor Deficits in an Alpha-Synuclein Parkinson’s Model. Int. J. Mol. Sci. 2023, 24, 4204. [Google Scholar] [CrossRef]
- Liu, Q.; Emadi, S.; Shen, J.-X.; Sierks, M.R.; Wu, J. Human A4β2 Nicotinic Acetylcholine Receptor as a Novel Target of Oligomeric α-Synuclein. PLoS ONE 2013, 8, e55886. [Google Scholar] [CrossRef]
- Ono, K.; Hirohata, M.; Yamada, M. Anti-Fibrillogenic and Fibril-Destabilizing Activity of Nicotine in Vitro: Implications for the Prevention and Therapeutics of Lewy Body Diseases. Exp. Neurol. 2007, 205, 414–424. [Google Scholar] [CrossRef]
- Ono, K.; Hirohata, M.; Yamada, M. Alpha-Synuclein Assembly as a Therapeutic Target of Parkinson’s Disease and Related Disorders. Curr. Pharm. Des. 2008, 14, 3247–3266. [Google Scholar] [CrossRef]
- Kardani, J.; Sethi, R.; Roy, I. Nicotine Slows down Oligomerisation of α-Synuclein and Ameliorates Cytotoxicity in a Yeast Model of Parkinson’s Disease. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 1454–1463. [Google Scholar] [CrossRef]
- Bono, F.; Mutti, V.; Savoia, P.; Barbon, A.; Bellucci, A.; Missale, C.; Fiorentini, C. Nicotine Prevents Alpha-Synuclein Accumulation in Mouse and Human IPSC-Derived Dopaminergic Neurons through Activation of the Dopamine D3- Acetylcholine Nicotinic Receptor Heteromer. Neurobiol. Dis. 2019, 129, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, Y.; Li, Y.; Xu, S.; Tao, T.; Hua, Y.; Zhang, J.; Fan, Y. Activation of A7-NAChRs Promotes the Clearance of α-Synuclein and Protects Against Apoptotic Cell Death Induced by Exogenous α-Synuclein Fibrils. Front. Cell Dev. Biol. 2021, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Elgayar, S.A.M.; Hussein, O.A.; Mubarak, H.A.; Ismaiel, A.M.; Gomaa, A.M.S. Testing Efficacy of the Nicotine Protection of the Substantia Nigra Pars Compacta in a Rat Parkinson Disease Model. Ultrastructure Study. Ultrastruct. Pathol. 2022, 46, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Kalsoom, I.; Wang, Y.; Li, B.; Wen, H. Research Progress of α-Synuclein Aggregation Inhibitors for Potential Parkinson’s Disease Treatment. Mini. Rev. Med. Chem. 2023, 23, 1959–1974. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Magen, I.; Bove, N.; Zhu, C.; Lemesre, V.; Dutta, G.; Elias, C.J.; Lester, H.A.; Chesselet, M.F. Chronic Nicotine Improves Cognitive and Social Impairment in Mice Overexpressing Wild Type α-Synuclein. Neurobiol. Dis. 2018, 117, 170–180. [Google Scholar] [CrossRef]
- Fan, T.-S.; Liu, S.C.-H.; Wu, R.-M. Alpha-Synuclein and Cognitive Decline in Parkinson Disease. Life 2021, 11, 1239. [Google Scholar] [CrossRef]
- Camilo Jurado-Coronel, J.; Avila-Rodriguez, M.; Capani, F.; Gonzalez, J.; Echeverria Moran, V.; Barreto, G.E. Targeting the Nicotinic Acetylcholine Receptors (NAChRs) in Astrocytes as a Potential Therapeutic Target in Parkinson’s Disease. Curr. Pharm. Des. 2016, 22, 1305–1311. [Google Scholar] [CrossRef]
- Olsen, A.L.; Clemens, S.G.; Feany, M.B. Nicotine-Mediated Rescue of α-Synuclein Toxicity Requires Synaptic Vesicle Glycoprotein 2 in Drosophila. Mov. Disord. 2023, 38, 244–255. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B. Nicotinic Receptor Intervention in Parkinson’s Disease: Future Directions. Clin. Pharmacol. Transl. Med. 2017, 1, 14. [Google Scholar]
- Mitra, S.; Khatri, S.N.; Maulik, M.; Bult-Ito, A.; Schulte, M. Allosterism of Nicotinic Acetylcholine Receptors: Therapeutic Potential for Neuroinflammation Underlying Brain Trauma and Degenerative Disorders. Int. J. Mol. Sci. 2020, 21, 4918. [Google Scholar] [CrossRef]
- Manetti, D.; Dei, S.; Arias, H.R.; Braconi, L.; Gabellini, A.; Teodori, E.; Romanelli, M.N. Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators. Molecules 2023, 28, 1270. [Google Scholar] [CrossRef] [PubMed]
- Sanders, V.R.; Millar, N.S. Potentiation and Allosteric Agonist Activation of α7 Nicotinic Acetylcholine Receptors: Binding Sites and Hypotheses. Pharmacol. Res. 2023, 191, 106759. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The Cholinergic Anti-Inflammatory Pathway: A Missing Link in Neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B. Cholinergic Modulation of the Immune System Presents New Approaches for Treating Inflammation. Pharmacol. Ther. 2017, 179, 1–16. [Google Scholar] [CrossRef]
- Roa-Vidal, N.; Rodríguez-Aponte, A.S.; Lasalde-Dominicci, J.A.; Capó-Vélez, C.M.; Delgado-Vélez, M. Cholinergic Polarization of Human Macrophages. Int. J. Mol. Sci. 2023, 24, 15732. [Google Scholar] [CrossRef]
- Tracey, K.J. Fat Meets the Cholinergic Antiinflammatory Pathway. J. Exp. Med. 2005, 202, 1017–1021. [Google Scholar] [CrossRef]
- Rueda Ruzafa, L.; Cedillo, J.L.; Hone, A.J. Nicotinic Acetylcholine Receptor Involvement in Inflammatory Bowel Disease and Interactions with Gut Microbiota. Int. J. Environ. Res. Public. Health 2021, 18, 1189. [Google Scholar] [CrossRef]
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10, 1838. [Google Scholar] [CrossRef]
- Cavaliere, G.; Traina, G. Neuroinflammation in the Brain and Role of Intestinal Microbiota: An Overview of the Players. J. Integr. Neurosci. 2023, 22, 148. [Google Scholar] [CrossRef]
- Bostick, J.W.; Schonhoff, A.M.; Mazmanian, S.K. Gut Microbiome-Mediated Regulation of Neuroinflammation. Curr. Opin. Immunol. 2022, 76, 102177. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Interaction of Heavy Metal Lead with Gut Microbiota: Implications for Autism Spectrum Disorder. Biomolecules 2023, 13, 1549. [Google Scholar] [CrossRef] [PubMed]
- Claudino dos Santos, J.C.; Lima, M.P.P.; Brito, G.A.d.C.; Viana, G.S.d.B. Role of Enteric Glia and Microbiota-Gut-Brain Axis in Parkinson Disease Pathogenesis. Ageing Res. Rev. 2023, 84, 101812. [Google Scholar] [CrossRef] [PubMed]
- Pan, I.; Issac, P.K.; Rahman, M.d.M.; Guru, A.; Arockiaraj, J. Gut-Brain Axis a Key Player to Control Gut Dysbiosis in Neurological Diseases. Mol. Neurobiol. 2023, 1–19, online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Sokratian, A.; Chavez, K.R.; King, S.; Swain, S.M.; Snyder, J.C.; West, A.B.; Liddle, R.A. Gut Mucosal Cells Transfer α-Synuclein to the Vagus Nerve. JCI Insight 2023, 8, e172192. [Google Scholar] [CrossRef]
- Longo, S.; Rizza, S.; Federici, M. Microbiota-Gut-Brain Axis: Relationships among the Vagus Nerve, Gut Microbiota, Obesity, and Diabetes. Acta Diabetol. 2023, 60, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Takbiri Osgoei, L.; Parivar, K.; Ebrahimi, M.; Mortaz, E. Nicotine modulates the release of inflammatory cytokines and expression of TLR2, TLR4 of cord blood mononuclear cells. Iran. J. Allergy Asthma Immunol. 2018, 17, 372–378. [Google Scholar] [CrossRef]
- Randall, C.A.; Sun, D.; Randall, P.A. Differential Effects of Nicotine, Alcohol, and Coexposure on Neuroimmune-Related Protein and Gene Expression in Corticolimbic Brain Regions of Rats. ACS Chem. Neurosci. 2023, 14, 628–644. [Google Scholar] [CrossRef]
- Takata, K.; Kimura, H.; Yanagisawa, D.; Harada, K.; Nishimura, K.; Kitamura, Y.; Shimohama, S.; Tooyama, I. Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease. Molecules 2022, 27, 2780. [Google Scholar] [CrossRef]
- Zhang, X.; Lao, K.; Qiu, Z.; Rahman, M.S.; Zhang, Y.; Gou, X. Potential Astrocytic Receptors and Transporters in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 1109–1122. [Google Scholar] [CrossRef]
- Mizrachi, T.; Vaknin-Dembinsky, A.; Brenner, T.; Treinin, M. Neuroinflammation Modulation via α7 Nicotinic Acetylcholine Receptor and Its Chaperone, RIC-3. Molecules 2021, 26, 6139. [Google Scholar] [CrossRef]
- Fontana, I.C.; Kumar, A.; Nordberg, A. The Role of Astrocytic α7 Nicotinic Acetylcholine Receptors in Alzheimer Disease. Nat. Rev. Neurol. 2023, 19, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Oswald, M.J.; Han, Y.; Li, H.; Marashli, S.; Oglo, D.N.; Ojha, B.; Naser, P.V.; Gan, Z.; Kuner, R. Cholinergic Basal Forebrain Nucleus of Meynert Regulates Chronic Pain-like Behavior via Modulation of the Prelimbic Cortex. Nat. Commun. 2022, 13, 5014. [Google Scholar] [CrossRef]
- Koulousakis, P.; Andrade, P.; Visser-Vandewalle, V.; Sesia, T. The Nucleus Basalis of Meynert and Its Role in Deep Brain Stimulation for Cognitive Disorders: A Historical Perspective. J. Alzheimer’s Dis. 2019, 69, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; McIntire, J.; Ryan, S.; Dunah, A.; Loring, R. Anti-Inflammatory Effects of Astroglial α7 Nicotinic Acetylcholine Receptors Are Mediated by Inhibition of the NF-ΚB Pathway and Activation of the Nrf2 Pathway. J. Neuroinflammation 2017, 14, 192. [Google Scholar] [CrossRef]
- Piovesana, R.; Salazar Intriago, M.S.; Dini, L.; Tata, A.M. Cholinergic Modulation of Neuroinflammation: Focus on α7 Nicotinic Receptor. Int. J. Mol. Sci. 2021, 22, 4912. [Google Scholar] [CrossRef]
- Chen, J.-F.; Liu, K.; Hu, B.; Li, R.-R.; Xin, W.; Chen, H.; Wang, F.; Chen, L.; Li, R.-X.; Ren, S.-Y.; et al. Enhancing Myelin Renewal Reverses Cognitive Dysfunction in a Murine Model of Alzheimer’s Disease. Neuron 2021, 109, 2292–2307.e5. [Google Scholar] [CrossRef]
- Mazuir, E.; Fricker, D.; Sol-Foulon, N. Neuron–Oligodendrocyte Communication in Myelination of Cortical GABAergic Cells. Life 2021, 11, 216. [Google Scholar] [CrossRef]
- Thornton, M.A.; Hughes, E.G. Neuron-Oligodendroglia Interactions: Activity-Dependent Regulation of Cellular Signaling. Neurosci. Lett. 2020, 727, 134916. [Google Scholar] [CrossRef]
- Deng, S.; Shu, S.; Zhai, L.; Xia, S.; Cao, X.; Li, H.; Bao, X.; Liu, P.; Xu, Y. Optogenetic Stimulation of MPFC Alleviates White Matter Injury-Related Cognitive Decline after Chronic Ischemia through Adaptive Myelination. Adv. Sci. 2023, 10, 2202976. [Google Scholar] [CrossRef]
- Nagy, B.; Hovhannisyan, A.; Barzan, R.; Chen, T.-J.; Kukley, M. Different Patterns of Neuronal Activity Trigger Distinct Responses of Oligodendrocyte Precursor Cells in the Corpus Callosum. PLoS Biol. 2017, 15, e2001993. [Google Scholar] [CrossRef]
- Maas, D.A.; Angulo, M.C. Can Enhancing Neuronal Activity Improve Myelin Repair in Multiple Sclerosis? Front. Cell Neurosci. 2021, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D.; Dutta, D.J.; Belgrad, J.; Robnett, M. Cholinergic Signaling in Myelination. Glia 2017, 65, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Imamura, O.; Arai, M.; Dateki, M.; Oishi, K.; Takishima, K. Donepezil-induced Oligodendrocyte Differentiation Is Mediated through Estrogen Receptors. J. Neurochem. 2020, 155, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Poggi, G.; Wennström, M.; Müller, M.B.; Treccani, G. NG2-Glia: Rising Stars in Stress-Related Mental Disorders? Mol. Psychiatry. 2023, 28, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.A.; Wallis, G.J.; Wilcox, K.S. Reactivity and Increased Proliferation of NG2 Cells Following Central Nervous System Infection with Theiler’s Murine Encephalomyelitis Virus. J. Neuroinflamm. 2020, 17, 369. [Google Scholar] [CrossRef]
- Janeckova, L.; Knotek, T.; Kriska, J.; Hermanova, Z.; Kirdajova, D.; Kubovciak, J.; Berkova, L.; Tureckova, J.; Camacho Garcia, S.; Galuskova, K.; et al. Astrocyte-like Subpopulation of NG2 Glia in the Adult Mouse Cortex Exhibits Characteristics of Neural Progenitor Cells. Glia 2024, 72, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Inden, M.; Minamino, H.; Abe, M.; Takata, K.; Taniguchi, T. The 6-Hydroxydopamine-Induced Nigrostriatal Neurodegeneration Produces Microglia-like NG2 Glial Cells in the Rat Substantia Nigra. Glia 2010, 58, 1686–1700. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Sun, T.; Li, M.; Liu, Q.; Yu, A.; Cheng, K.; Ma, J.; Murphy, S.; McNutt, P.M.; Zhang, Y. Insights into Optimizing Exosome Therapies for Acute Skin Wound Healing and Other Tissue Repair. Front. Med. 2024, 1–27, epub ahead of print. [Google Scholar] [CrossRef]
- Tan, F.; Li, X.; Wang, Z.; Li, J.; Shahzad, K.; Zheng, J. Clinical Applications of Stem Cell-Derived Exosomes. Signal Transduct. Target. Ther. 2024, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, G.C.; Bortolanza, M.; Bribian, A.; Leal-Luiz, G.C.; Raisman-Vozari, R.; López-Mascaraque, L.; Del-Bel, E. Dynamic Involvement of Striatal NG2-Glia in L-DOPA Induced Dyskinesia in Parkinsonian Rats: Effects of Doxycycline. ASN Neuro 2023, 15, 175909142311559. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Matsubara, K.; Matsubara, Y.; Nakaoka, H.; Sugiyama, T. The Influence of Nicotine on Trophoblast-Derived Exosomes in a Mouse Model of Pathogenic Preeclampsia. Int. J. Mol. Sci. 2023, 24, 11126. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares, É.N.; Costa, A.C.d.S.; Ferrolho, G.d.J.; Ureshino, R.P.; Getachew, B.; Costa, S.L.; da Silva, V.D.A.; Tizabi, Y. Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease. Cells 2024, 13, 474. https://doi.org/10.3390/cells13060474
Soares ÉN, Costa ACdS, Ferrolho GdJ, Ureshino RP, Getachew B, Costa SL, da Silva VDA, Tizabi Y. Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease. Cells. 2024; 13(6):474. https://doi.org/10.3390/cells13060474
Chicago/Turabian StyleSoares, Érica Novaes, Ana Carla dos Santos Costa, Gabriel de Jesus Ferrolho, Rodrigo Portes Ureshino, Bruk Getachew, Silvia Lima Costa, Victor Diogenes Amaral da Silva, and Yousef Tizabi. 2024. "Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease" Cells 13, no. 6: 474. https://doi.org/10.3390/cells13060474