
Progranulin Protects against Hyperglycemia-Induced Neuronal Dysfunction through GSK3β Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Cell Culture
2.2. Hyperglycemia, Progranulin, and Inhibitor Treatment
2.3. Determination of Cell Viability and Neurite Outgrowth
2.4. Determination of Mitochondrial Viability
2.5. Mitochondrial Complex Enzyme Activity Assays
2.6. Immunofluorescence
2.7. Immunoblotting
2.8. Calcium Imaging
2.9. Multielectrode Array (MEA)
2.10. Statistical Analysis
3. Results
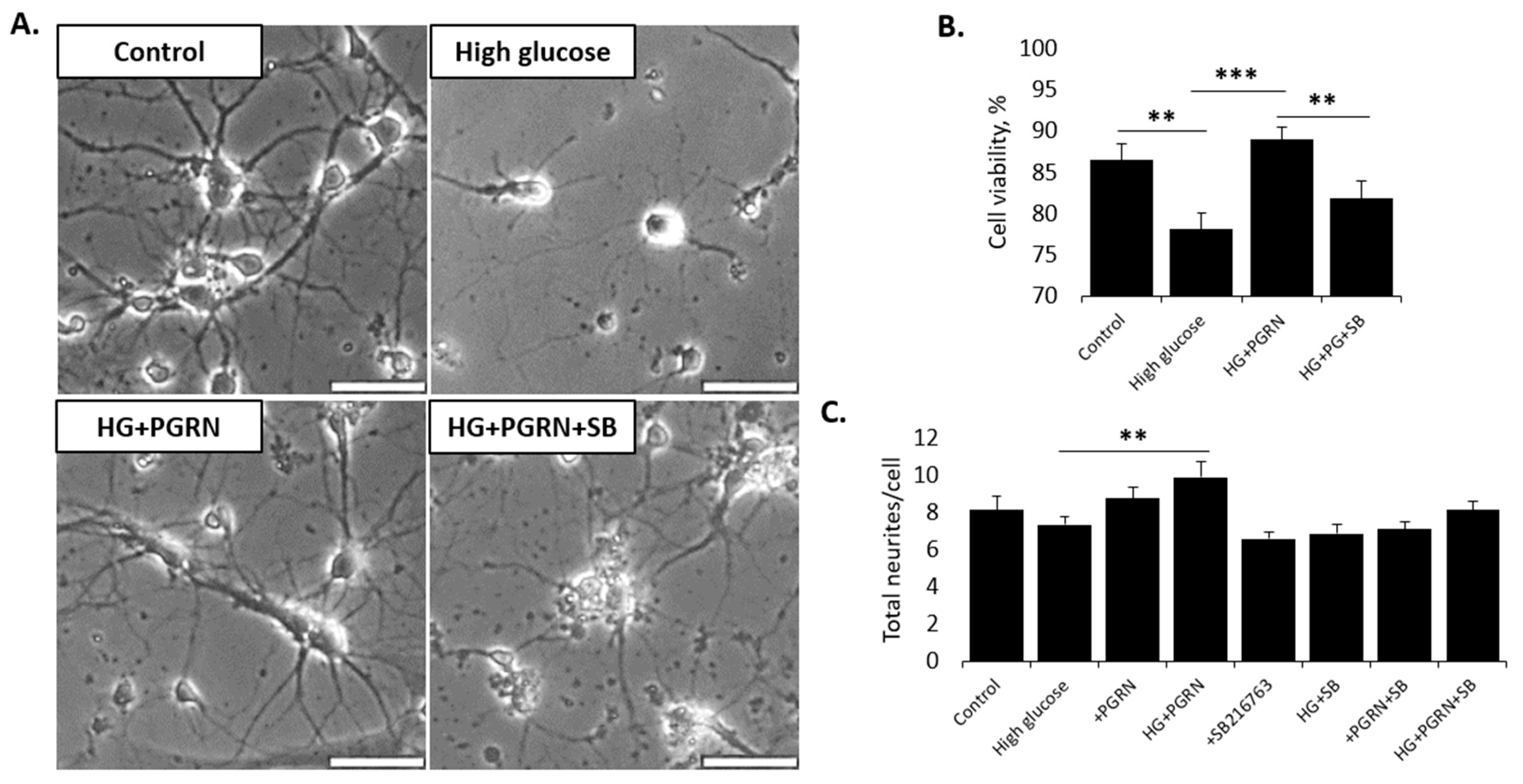
3.1. Neuronal Cell Viability under High-Glucose Stress Is Preserved by PGRN in a GSK3β-Dependent Manner
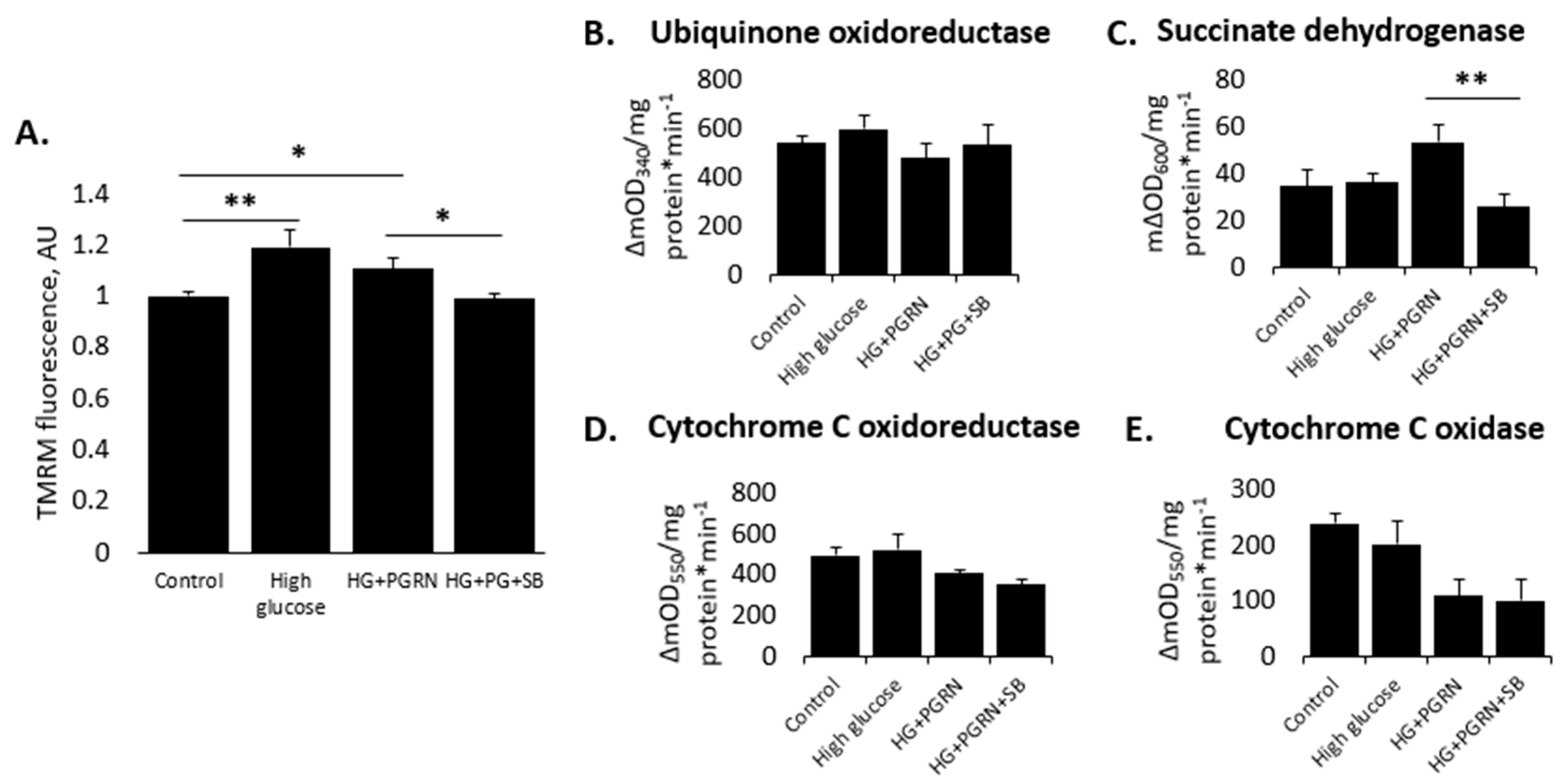
3.2. Neuronal Mitochondrial Health and Activity Is Preserved by PGRN in a GSK3β-Dependent Manner under High-Glucose Stress
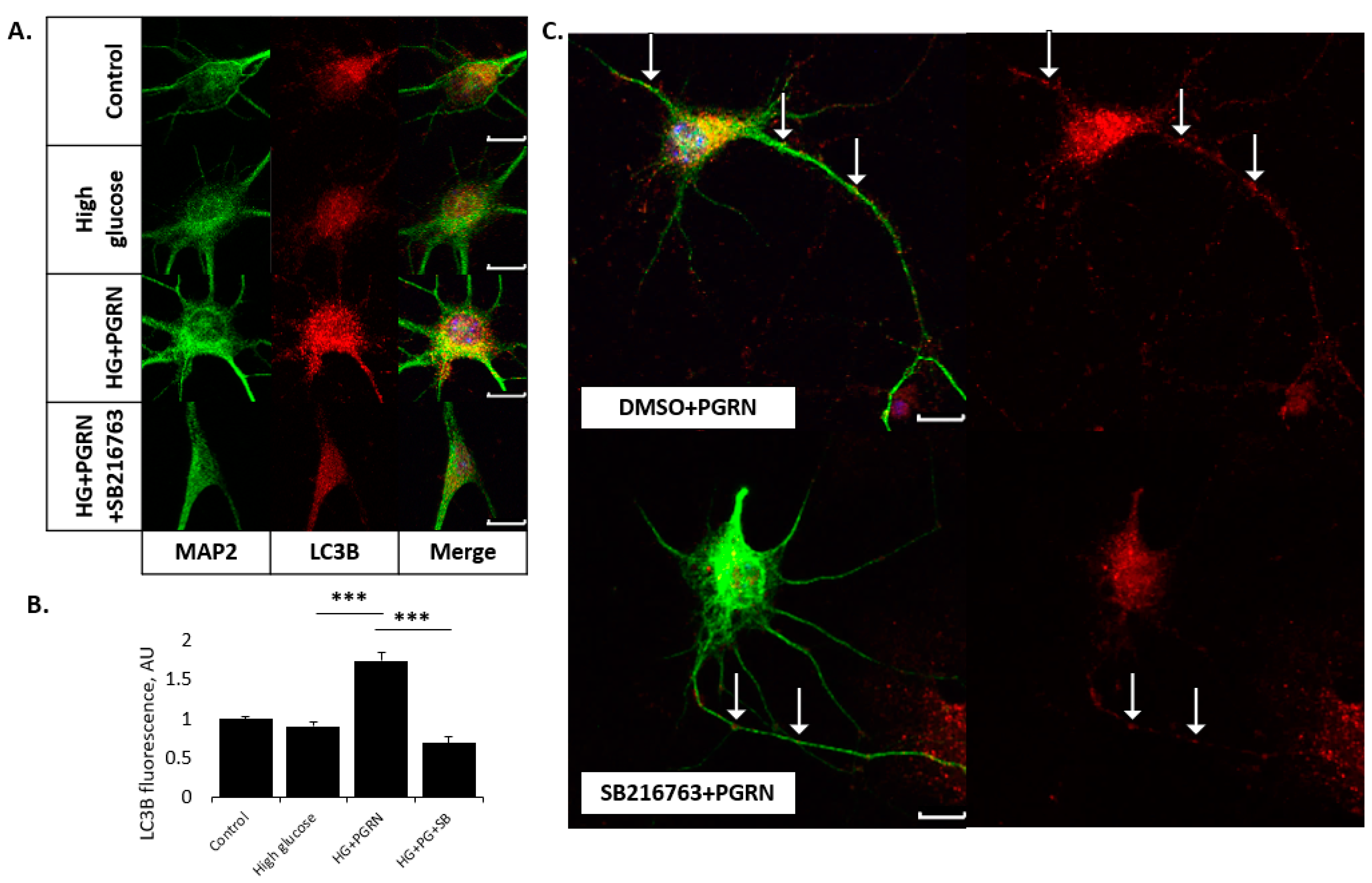
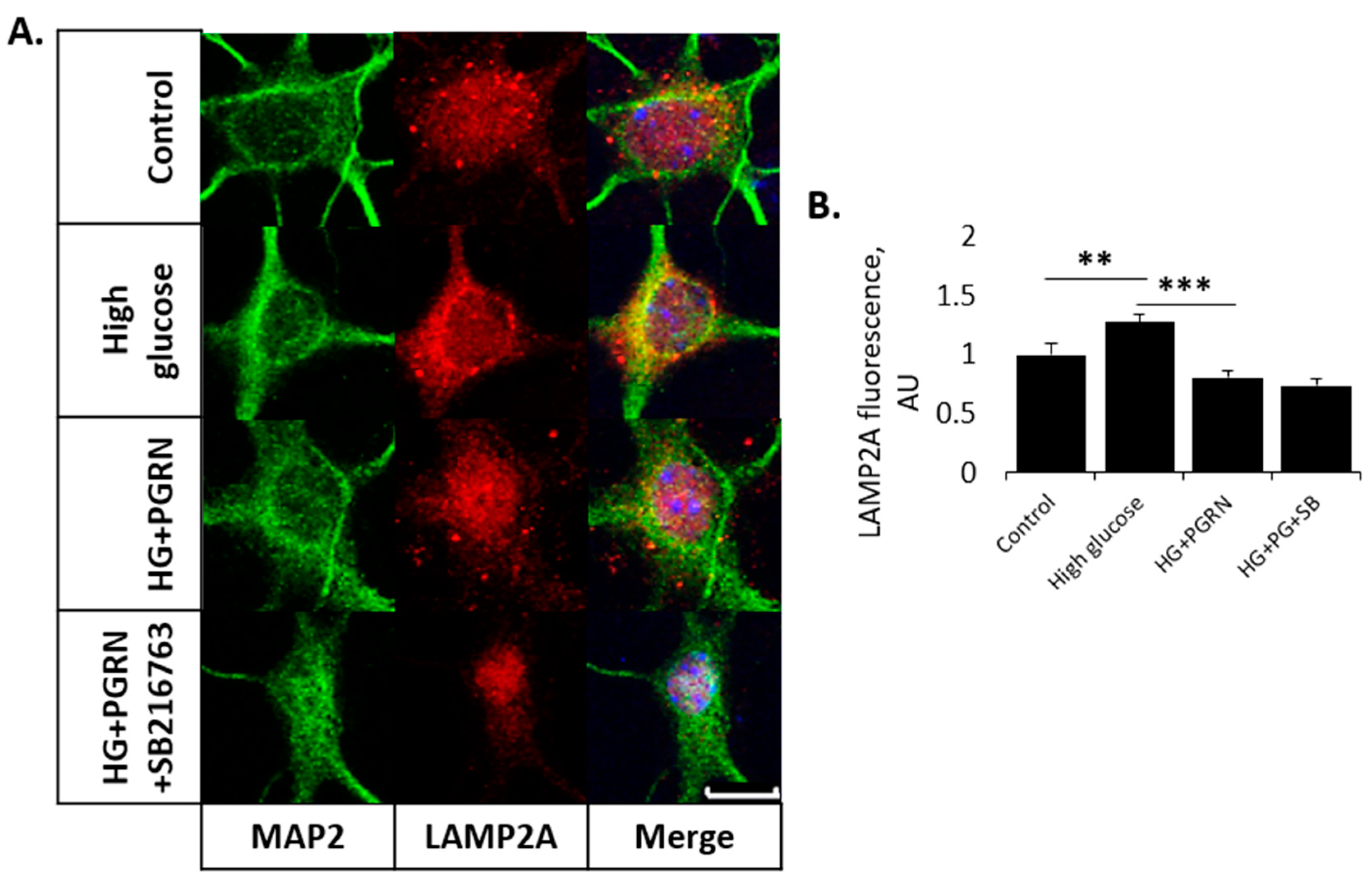
3.3. PGRN-Mediated Autophagy Flux under High Glucose Involves GSK3β Activity
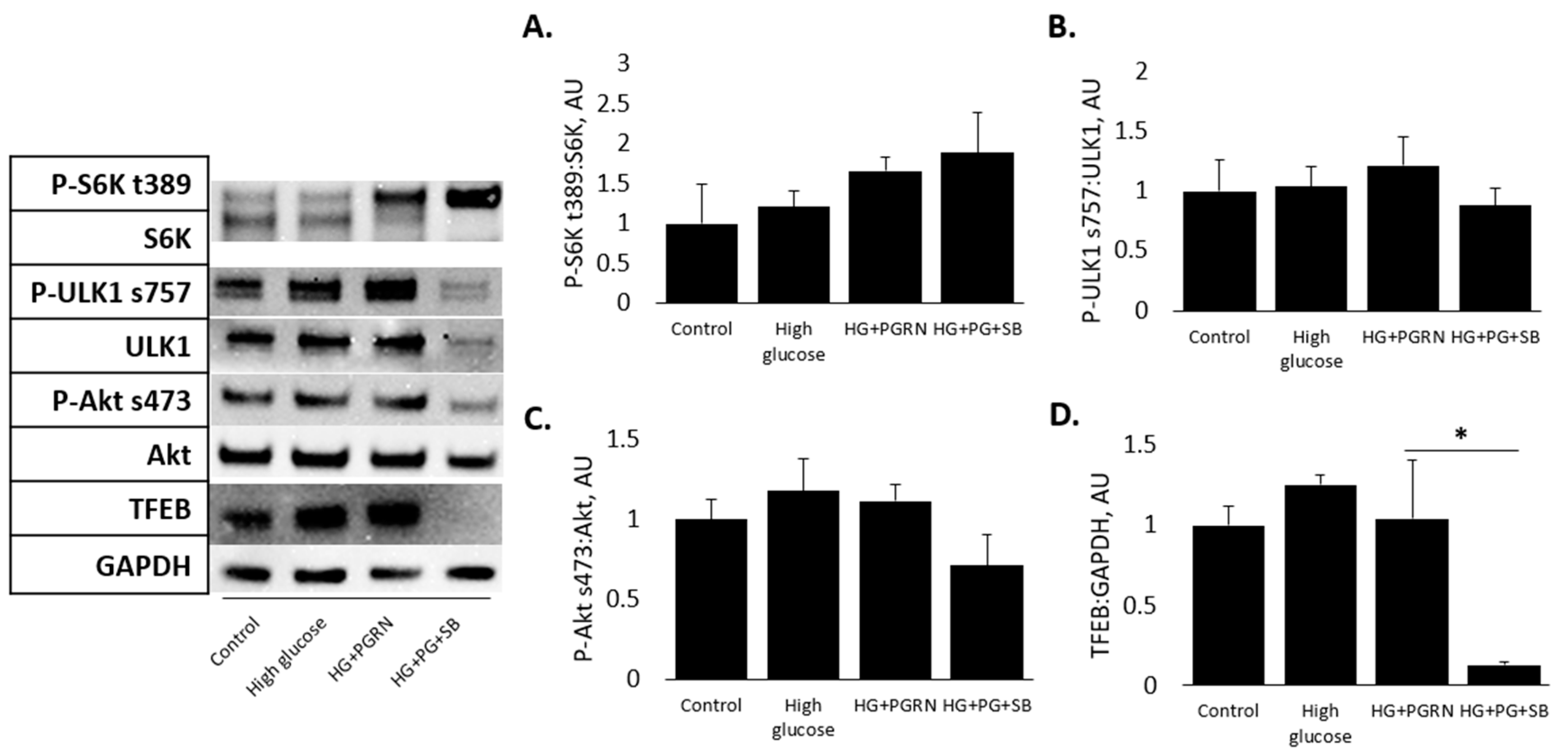
3.4. mTOR and Akt Pathway Phosphorylation Do Not Appear to Be Affected by PGRN under High Glucose in Cortical Cells
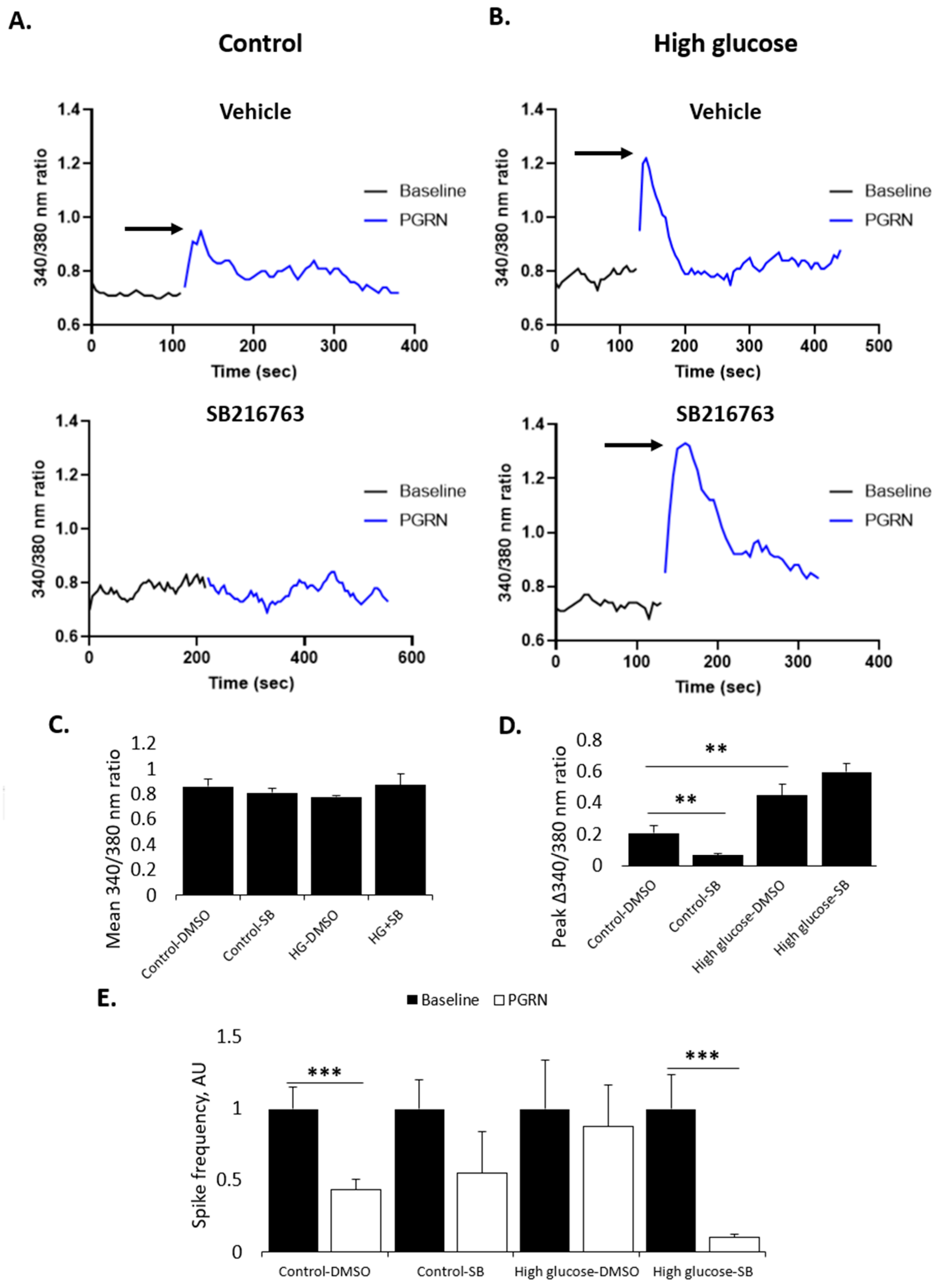
3.5. PGRN-Mediated Changes in Calcium Influx and Neuronal Firing Are Affected by GSK3β
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bommer, C.; Heesemann, E.; Sagalova, V.; Manne-Goehler, J.; Atun, R.; Bärnighausen, T.; Vollmer, S. The global economic burden of diabetes in adults aged 20–79 years: A cost-of-illness study. Lancet Diabetes Endocrinol. 2017, 5, 423–430. [Google Scholar] [CrossRef]
- Dewanjee, S.; Das, S.; Das, A.K.; Bhattacharjee, N.; Dihingia, A.; Dua, T.K.; Kalita, J.; Manna, P. Molecular mechanism of diabetic neuropathy and its pharmacotherapeutic targets. Eur. J. Pharmacol. 2018, 833, 472–523. [Google Scholar] [CrossRef]
- Sherling, D.H.; Perumareddi, P.; Hennekens, C.H. Metabolic Syndrome: Clinical and Policy Implications of the New Silent Killer. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. 2017, 59, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes–Evidence Reviewed. J. Diabetes Sci. Technol. Online 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergi, D.; Renaud, J.; Simola, N.; Martinoli, M.-G. Diabetes, a Contemporary Risk for Parkinson’s Disease: Epidemiological and Cellular Evidences. Front. Aging Neurosci. 2019, 11, 302. [Google Scholar] [CrossRef] [Green Version]
- Cassano, V.; Leo, A.; Tallarico, M.; Nesci, V.; Cimellaro, A.; Fiorentino, T.V.; Citraro, R.; Hribal, M.L.; De Sarro, G.; Perticone, F.; et al. Metabolic and Cognitive Effects of Ranolazine in Type 2 Diabetes Mellitus: Data from an in vivo Model. Nutrients 2020, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function via glucocorticoid–mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Zochodne, D.W.; Verge, V.M.K.; Cheng, C.; Sun, H.; Johnston, J. Does diabetes target ganglion neurones? Progressive sensory neurone involvement in long-term experimental diabetes. Brain 2001, 124, 2319–2334. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Mukhopadhyay, M.; Bhattacharyya, M.; Karmakar, P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J. 2018, 17, 709–720. [Google Scholar]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Son, J.H.; Shim, J.H.; Kim, K.-H.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein Turnover Via Autophagy: Implications for Metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito-Cuesta, I.; Ordóñez-Gutiérrez, L.; Wandosell, F. AMPK activation does not enhance autophagy in neurons in contrast to MTORC1 inhibition: Different impact on β-amyloid clearance. Autophagy 2020, 17, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Guan, K.-L.; Kim, J. AMPK and autophagy in glucose/glycogen metabolism. Mol. Asp. Med. 2015, 46, 46–62. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.-M.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 Signaling Pathway Links Growth Factor Deprivation to Autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef]
- Kao, A.W.; McKay, A.; Singh, P.P.; Brunet, A.; Huang, E.J. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 2017, 18, 325–333. [Google Scholar] [CrossRef]
- Daniel, R.; He, Z.; Carmichael, K.P.; Halper, J.; Bateman, A. Cellular Localization of Gene Expression for Progranulin. J. Histochem. Cytochem. 2000, 48, 999–1009. [Google Scholar] [CrossRef]
- Almeida, M.R.; Macário, M.C.; Ramos, L.; Baldeiras, I.; Ribeiro, M.H.; Santana, I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol. Aging 2016, 41, 200.e1–200.e5. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; Van Der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.-J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.; Cannon, A.; Mackenzie, I.R.; Boeve, B.; Baker, M.; Adamson, J.; Crook, R.; Melquist, S.; Kuntz, K.; Petersen, R.; et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006, 15, 2988–3001. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.; Sheng, H.; Xu, Y.-F.; Lin, W.-L.; Innes, A.E.; Gass, J.; Yu, X.; Hou, H.; Chiba, S.; Yamanouchi, K.; et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010, 177, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Gaweda-Walerych, K.; Walerych, D.; Berdyński, M.; Buratti, E.; Zekanowski, C. Parkin Levels Decrease in Fibroblasts With Progranulin (PGRN) Pathogenic Variants and in a Cellular Model of PGRN Deficiency. Front. Mol. Neurosci. 2021, 14, 676478. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Min, S.-W.; Krabbe, G.; Wang, C.; Zhou, Y.; Asgarov, R.; Li, Y.; Martens, L.H.; Elia, L.P.; E Ward, M.; et al. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 2014, 20, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Gass, J.; Lee, W.C.; Cook, C.; Finch, N.; Stetler, C.; Jansen-West, K.; Lewis, J.; Link, C.D.; Rademakers, R.; Nykjær, A.; et al. Progranulin regulates neuronal outgrowth independent of Sortilin. Mol. Neurodegener. 2012, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedert, C.; Mishra, V.; Aggarwal, G.; Nguyen, A.D.; Xu, F. Progranulin Preserves Autophagy Flux and Mitochondrial Function in Rat Cortical Neurons under High Glucose Stress. Front. Cell Neurosci. 2022, 16, 874258. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, J.; Hu, Y.; Liao, A.; Zheng, W.; Wang, X.; Lan, J.; Shen, J.; Wang, S.; Yang, F.; et al. Progranulin improves neural development via the PI3K/Akt/GSK-3β pathway in the cerebellum of a VPA-induced rat model of ASD. Transl. Psychiatry 2022, 12, 114. [Google Scholar] [CrossRef]
- Horinokita, I.; Hayashi, H.; Nagatomo, T.; Fushiki, Y.; Iwatani, Y.; Takagi, N. Effect of Progranulin on Proliferation and Differentiation of Neural Stem/Progenitor Cells after Oxygen/Glucose Deprivation. Int. J. Mol. Sci. 2022, 23, 1949. [Google Scholar] [CrossRef]
- Nedachi, T.; Kawai, T.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Progranulin enhances neural progenitor cell proliferation through glycogen synthase kinase 3β phosphorylation. Neuroscience 2011, 185, 106–115. [Google Scholar] [CrossRef]
- Kaeidi, A.; Hajializadeh, Z.; Jahandari, F.; Fatemi, I. Leptin attenuates oxidative stress and neuronal apoptosis in hyperglycemic condition. Fundam. Clin. Pharmacol. 2018, 33, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Cimen, H.; Han, M.-J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of Succinate Dehydrogenase Activity by SIRT3 in Mammalian Mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-Y.; Zhang, X.-L.; Wu, T.-F.; Liu, Y.-P.; Wang, Q.; Zhang, Y.; Song, J.-Q.; Wang, Y.-J.; Yang, Y.-L. Analysis of the Mitochondrial Complex I-V Enzyme Activities of Peripheral Leukocytes in Oxidative Phosphorylation Disorders. J. Child Neurol. 2011, 26, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ning, C.; Mu, J.; Li, D.; Ma, Y.; Meng, X. Role of Wnt signaling pathways in type 2 diabetes mellitus. Mol. Cell Biochem. 2021, 476, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Cox, L.R.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Piazza, F.; Manni, S.; Tubi, L.Q.; Montini, B.; Pavan, L.; Colpo, A.; Gnoato, M.; Cabrelle, A.; Adami, F.; Zambello, R.; et al. Glycogen Synthase Kinase-3 regulates multiple myeloma cell growth and bortezomib-induced cell death. BMC Cancer 2010, 10, 526. [Google Scholar] [CrossRef]
- Gebhardt, R.; Lerche, K.S.; Götschel, F.; Günther, R.; Kolander, J.; Teich, L.; Zellmer, S.; Hofmann, H.-J.; Eger, K.; Hecht, A.; et al. 4-Aminoethylamino-emodin—A novel potent inhibitor of GSK-3β–acts as an insulin-sensitizer avoiding downstream effects of activated β-catenin. J. Cell. Mol. Med. 2009, 14, 1276–1293. [Google Scholar] [CrossRef] [Green Version]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2009, 14, 1276–1293. [Google Scholar] [CrossRef]
- Cen, X.; Zhang, M.; Zhou, M.; Ye, L.; Xia, H. Mitophagy Regulates Neurodegenerative Diseases. Cells 2021, 10, 1876. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Tan, A.; Prasad, R.; Lee, C.; Jho, E.-H. Past, present, and future perspectives of transcription factor EB (TFEB): Mechanisms of regulation and association with disease. Cell Death Differ. 2022, 29, 1433–1449. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. BioMed Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [Green Version]
- Bautista, S.J.; Boras, I.; Vissa, A.; Mecica, N.; Yip, C.M.; Kim, P.K.; Antonescu, C.N. mTOR complex 1 controls the nuclear localization and function of glycogen synthase kinase 3β. J. Biol. Chem. 2018, 293, 14723–14739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 promotes metabolic reprogramming by suppression of GSK3-dependent Foxk1 phosphorylation. Mol. Cell 2018, 70, 949–960.e4. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, J.; Di, L. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef]
- Singh, J.N.; Negi, G.; Balbhim, S.; Mule, N.K.; Sharma, D.; Sharma, S.S. Short-term extracellular glucose exposure alters neurite outgrowth and intracellular reactive oxygen species without altering viability in neuronal cells. NISCAIR-CSIR 2017, 55, 648–654. [Google Scholar]
- Hur, E.-M.; Zhou, F.-Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.-Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.; Rimondini, R.; Viggiano, R.; Trazzi, S.; De Franceschi, M.; Bartesaghi, R.; Ciani, E. Inhibition of GSK3β rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 2015, 82, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Han, X.; Zou, H.; Zhang, B.; Ding, Y.; Xu, X.; Zeng, J.; Liu, J.; Gong, A. PTEN-GSK3β-MOB1 axis controls neurite outgrowth in vitro and in vivo. Cell Mol. Life Sci. CMLS 2018, 75, 4445–4464. [Google Scholar] [CrossRef] [PubMed]
- Alabed, Y.Z.; Pool, M.; Tone, S.O.; Sutherland, C.; Fournier, A.E. GSK3β Regulates Myelin-Dependent Axon Outgrowth Inhibition through CRMP4. J. Neurosci. 2010, 30, 5635–5643. [Google Scholar] [CrossRef] [Green Version]
- Morgan-Smith, M.; Wu, Y.; Zhu, X.; Pringle, J.; Snider, W.D. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. eLife 2014, 3, e02663. [Google Scholar] [CrossRef]
- Gao, X.; Joselin, A.P.; Wang, L.; Kar, A.; Ray, P.; Bateman, A.; Goate, A.M.; Wu, J.Y. Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3β. Protein Cell 2010, 1, 552–562. [Google Scholar] [CrossRef] [Green Version]
- Park, C.-H.; Lee, B.-H.; Ahn, S.-G.; Yoon, J.-H.; Oh, S.-H. Serine 9 and Tyrosine 216 Phosphorylation of GSK-3β Differentially Regulates Autophagy in Acquired Cadmium Resistance. Toxicol. Sci. 2013, 135, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Takabatake, Y.; Kimura, T.; Maejima, I.; Namba, T.; Yamamoto, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; et al. Autophagy Inhibits the Accumulation of Advanced Glycation End Products by Promoting Lysosomal Biogenesis and Function in the Kidney Proximal Tubules. Diabetes 2017, 66, 1359–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Nie, T.; Yang, S.; Ma, H.; Zhang, L.; Lu, F.; Tao, K.; Wang, R.; Yang, R.; Huang, L.; Mao, Z.; et al. Regulation of ER stress-induced autophagy by GSK3β-TIP60-ULK1 pathway. Cell Death Dis. 2016, 7, e2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Seol, D.-W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, S.; Minguzzi, M.; Platano, D.; Santi, S.; Trisolino, G.; Filardo, G.; Mariani, E.; Borzì, R.M. Glycogen Synthase Kinase-3β Inhibition Links Mitochondrial Dysfunction, Extracellular Matrix Remodelling and Terminal Differentiation in Chondrocytes. Sci. Rep. 2017, 7, 12059. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Wani, A.; Gupta, M.; Ahmad, M.; Shah, A.M.; Ahsan, A.U.; Qazi, P.H.; Malik, F.; Singh, G.; Sharma, P.R.; Kaddoumi, A.; et al. Alborixin clears amyloid-β by inducing autophagy through PTEN-mediated inhibition of the AKT pathway. Autophagy 2019, 15, 1810–1828. [Google Scholar] [CrossRef]
- Ways, D.; Sheetz, M.J. The role of protein kinase C in the development of the complications of diabetes. Vitam. Horm. 2000, 60, 149–193. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Mi, T.; Wang, R.; Zhang, Z.; Li, W.; Zhao, J.; Yang, P.; Xia, H.; Mao, Q. Progranulin deficiency promotes persistent neuroinflammation and causes regional pathology in the hippocampus following traumatic brain injury. Glia 2022, 70, 1317–1336. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Progranulin Protects Hippocampal Neurogenesis via Suppression of Neuroinflammatory Responses Under Acute Immune Stress. Mol. Neurobiol. 2016, 54, 3717–3728. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedert, C.; Salih, L.; Xu, F. Progranulin Protects against Hyperglycemia-Induced Neuronal Dysfunction through GSK3β Signaling. Cells 2023, 12, 1803. https://doi.org/10.3390/cells12131803
Dedert C, Salih L, Xu F. Progranulin Protects against Hyperglycemia-Induced Neuronal Dysfunction through GSK3β Signaling. Cells. 2023; 12(13):1803. https://doi.org/10.3390/cells12131803
Chicago/Turabian StyleDedert, Cass, Lyuba Salih, and Fenglian Xu. 2023. "Progranulin Protects against Hyperglycemia-Induced Neuronal Dysfunction through GSK3β Signaling" Cells 12, no. 13: 1803. https://doi.org/10.3390/cells12131803