
Evidence for Involvement of ADP-Ribosylation Factor 6 in Intracellular Trafficking and Release of Murine Leukemia Virus Gag

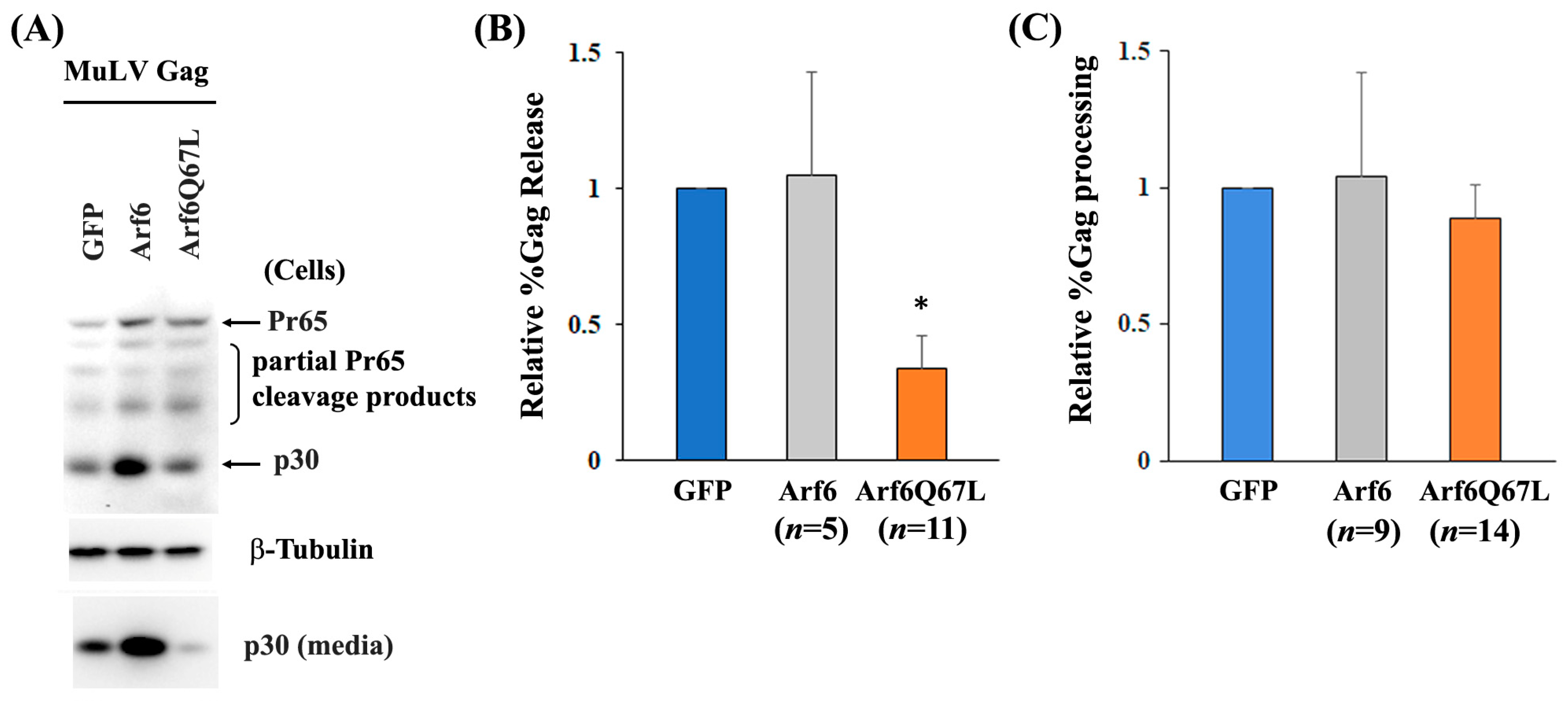{kind=link}
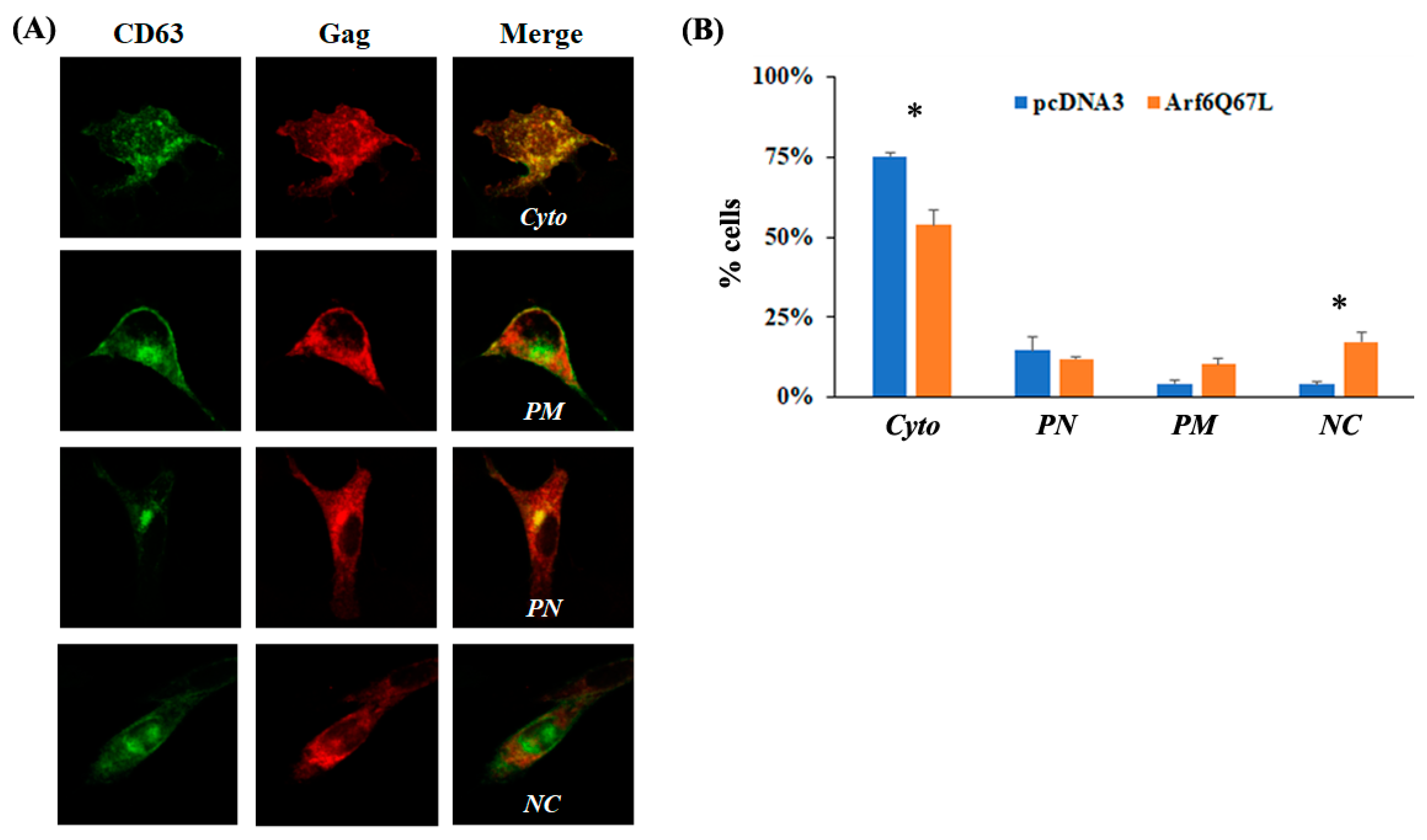{kind=link}
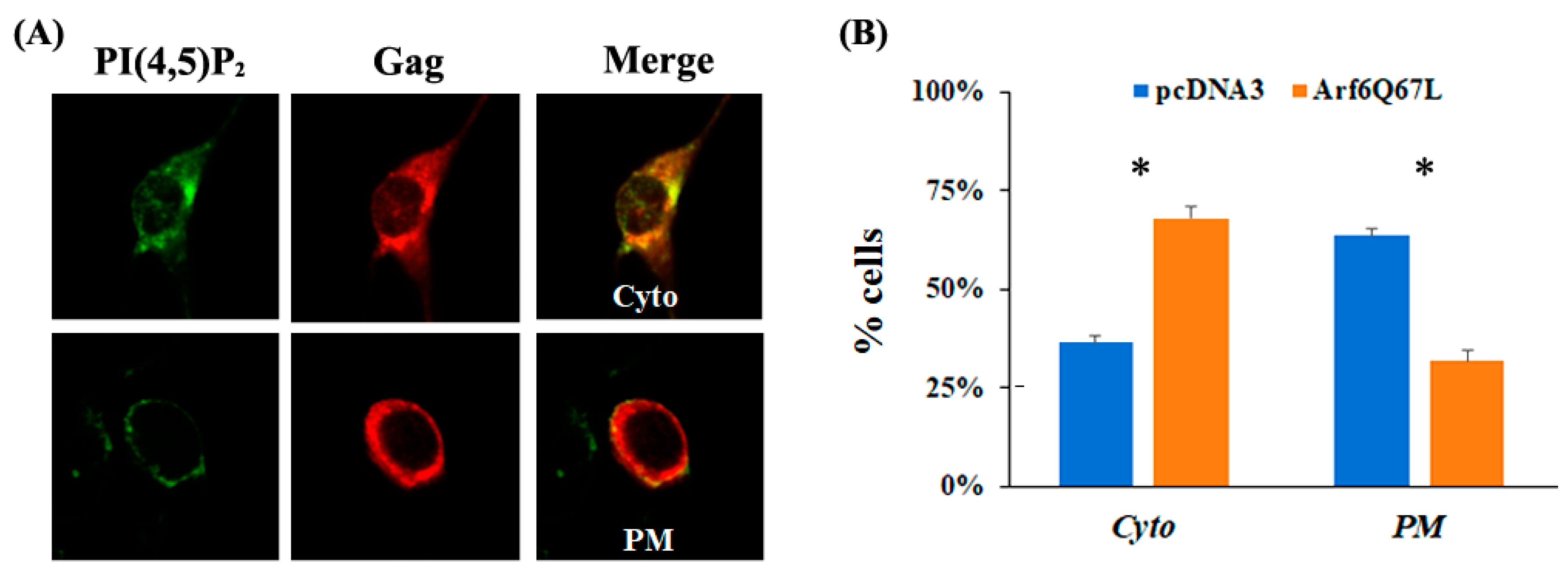{kind=link}
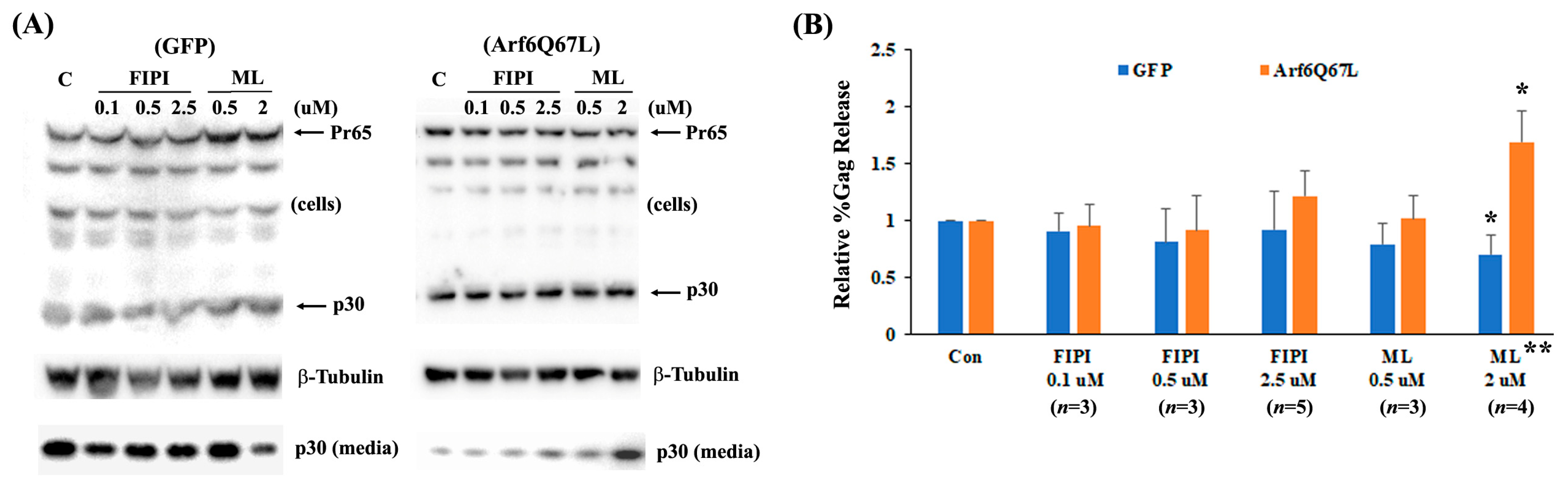{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Cell Culture
2.3. DNA Constructs
2.4. Indirect Immunofluorescence Microscopy
2.5. Virus Release Efficiency (%Gag Release)
2.6. Statistical Analysis
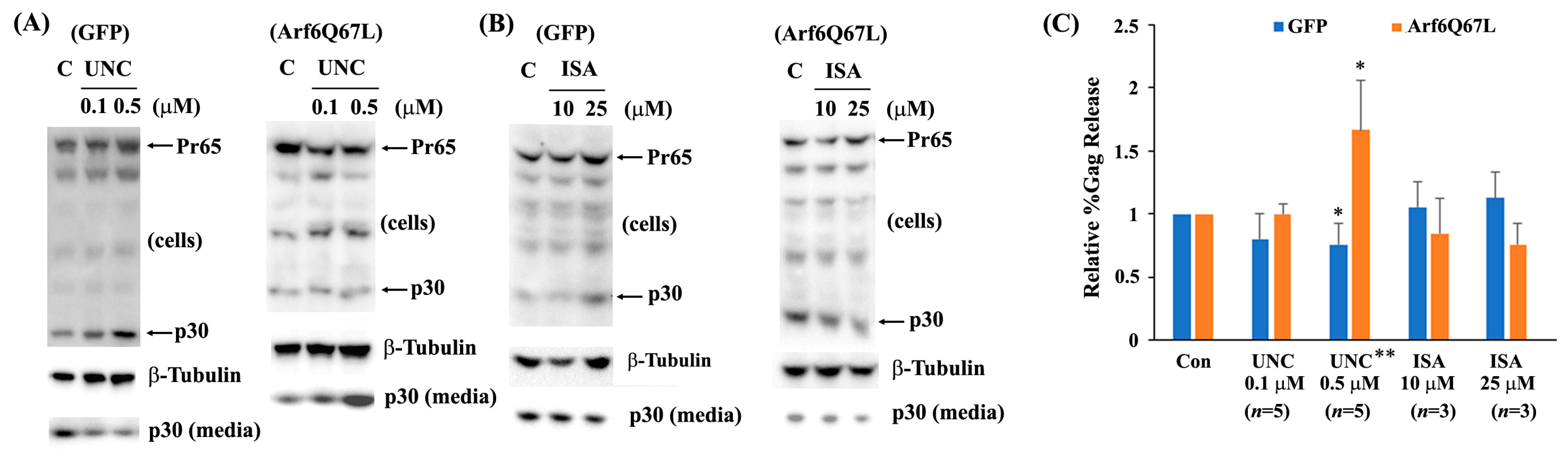
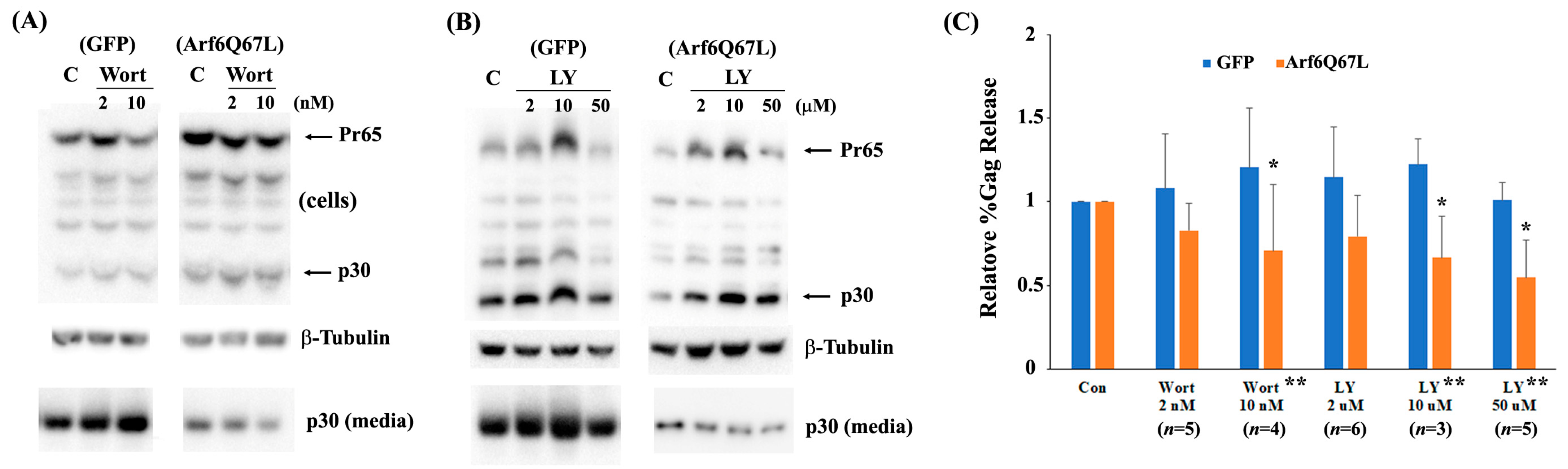
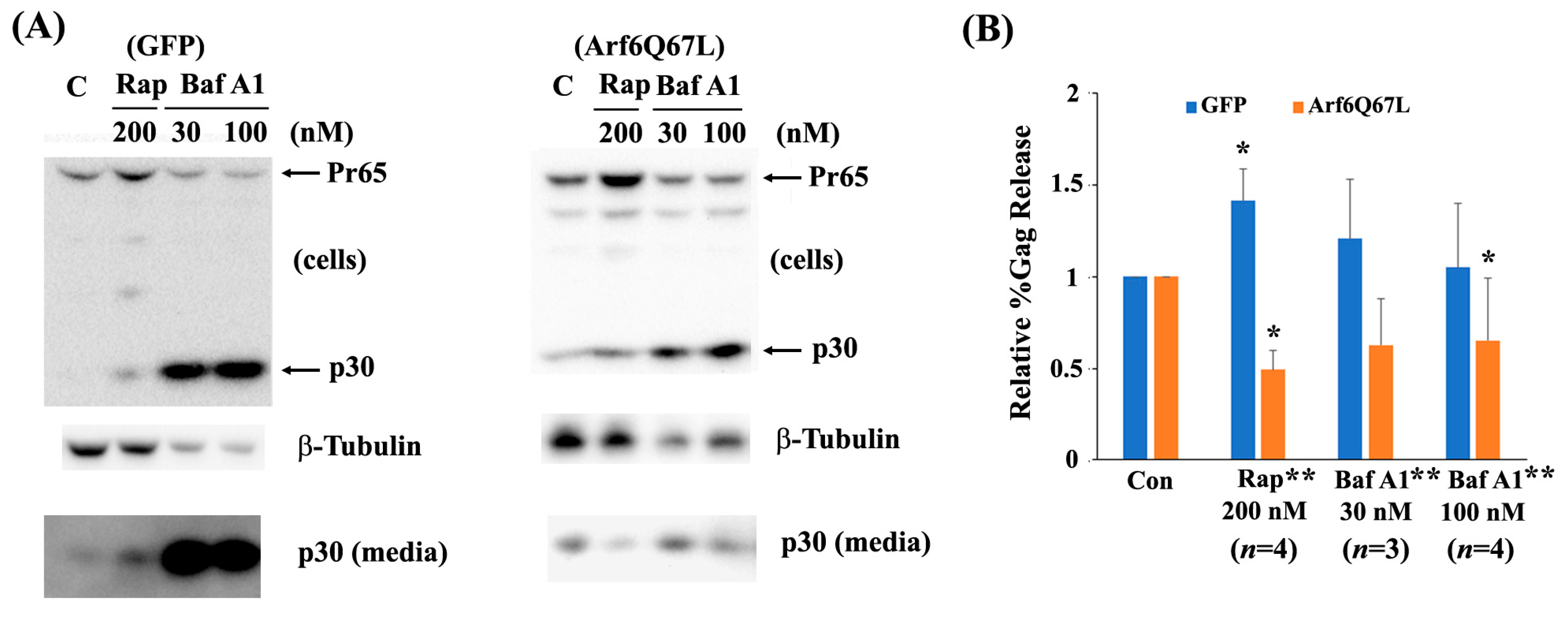
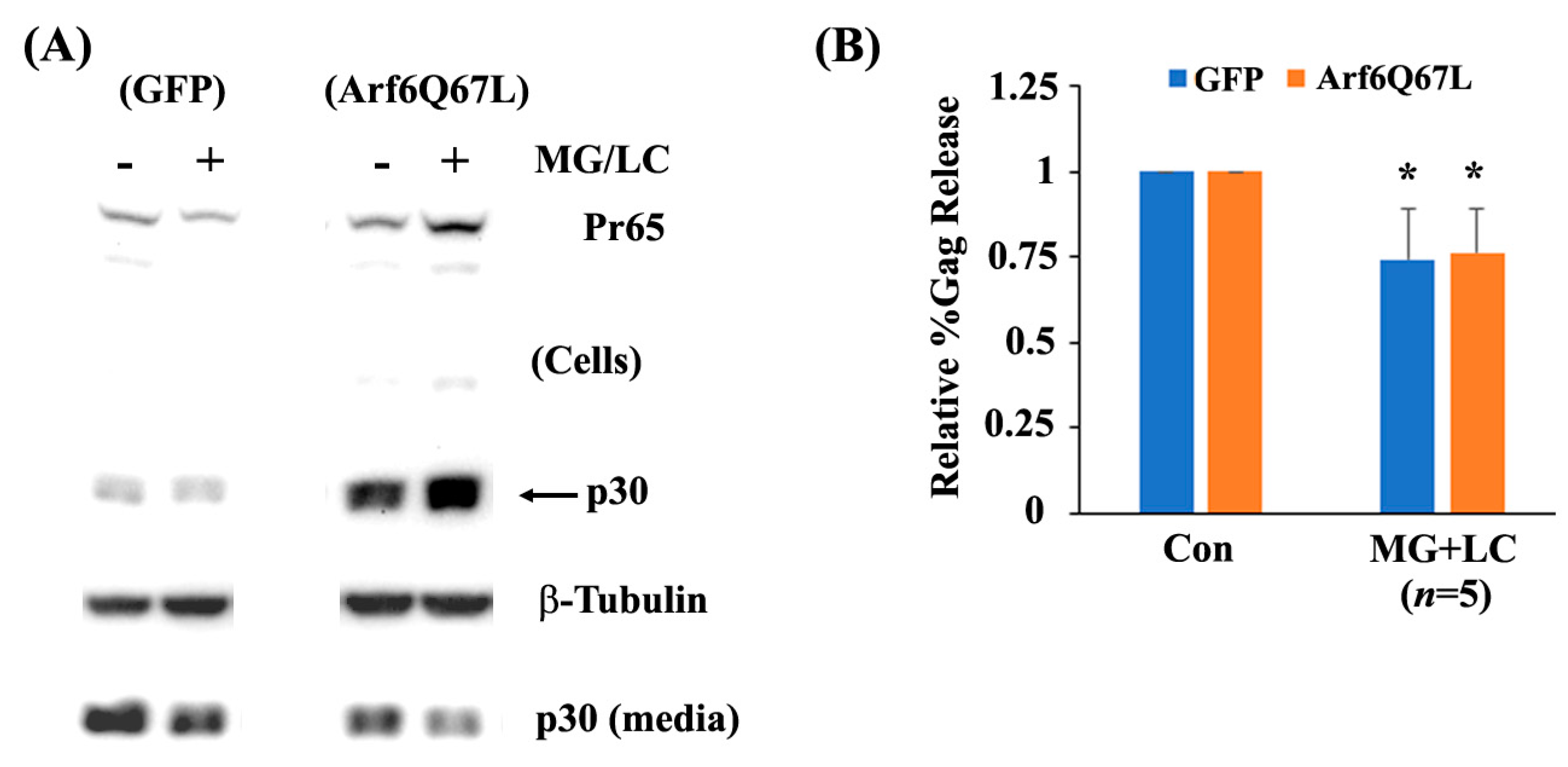
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rein, A. Murine Leukemia Viruses: Objects and Organisms. Adv. Virol. 2011, 2011, 403419. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Neil, S.J.D. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 2011, 9, 519–531. [Google Scholar] [CrossRef]
- Van Acker, T.; Tavernier, J.; Peelman, F. The Small GTPase Arf6: An Overview of Its Mechanisms of Action and of Its Role in Host–Pathogen Interactions and Innate Immunity. Int. J. Mol. Sci. 2019, 20, 2209. [Google Scholar] [CrossRef] [PubMed]
- Adarska, P.; Wong-Dilworth, L.; Bottanelli, F. ARF GTPases and Their Ubiquitous Role in Intracellular Trafficking Beyond the Golgi. Front. Cell Dev. Biol. 2021, 9, 679046. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, E.; Tran Nguyen, A.P.; Kassas, N.; Bader, M.F.; Grant, N.J.; Vitale, N. Regulation of Phospholipase D by Arf6 during FcgammaR-Mediated Phagocytosis. J. Immunol. 2019, 202, 2971–2981. [Google Scholar] [CrossRef]
- Funakoshi, Y.; Hasegawa, H.; Kanaho, Y. Regulation of PIP5K activity by Arf6 and its physiological significance. J. Cell. Physiol. 2011, 226, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Santy, L.C.; Casanova, J.E. Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J. Cell Biol. 2001, 154, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef]
- Brown, H.; Gutowski, S.; Moomaw, C.R.; Slaughter, C.; Sternwels, P.C. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 1993, 75, 1137–1144. [Google Scholar] [CrossRef]
- Moritz, A.; De Graan, P.; Gispen, W.; Wirtz, K. Phosphatidic acid is a specific activator of phosphatidylinositol-4-phosphate kinase. J. Biol. Chem. 1992, 267, 7207–7210. [Google Scholar] [CrossRef]
- Tan, X.; Thapa, N.; Liao, Y.; Choi, S.; Anderson, R.A. PtdIns(4,5)P2 signaling regulates ATG14 and autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Ravikumar, B.; Puri, C.; Rubinsztein, D.C. Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J. Cell Biol. 2012, 196, 483–496. [Google Scholar] [CrossRef]
- Mueller-Lantzsch, N.; Fan, H. Monospecific immunoprecipitation of murine leukemia virus polyribosomes: Identification of p30 protein-specific messenger RNA. Cell 1976, 9, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Tam, R.; Kim, J.W.; Fan, H. The Cellular Protein La Functions in Enhancement of Virus Release through Lipid Rafts Facilitated by Murine Leukemia Virus Glycosylated Gag. mBio 2011, 2, e00341-10. [Google Scholar] [CrossRef]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Kräusslich, H.-G.; Yewdell, J.W. Proteasome inhibition interferes with Gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.; Short, S.M.; Subramanian, R.R.; Zetter, B.R.; Roberts, T.M. DEF-1/ASAP1 is a GTPase-activating protein (GAP) for ARF1 that enhances cell motility through a GAP-dependent mechanism. J. Biol. Chem. 2002, 277, 7962–7969. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H.; Bannert, K.; Berlin, P.; Reiner, J.; Lemcke, H.; David, R.; Engelmann, R.; Lamprecht, G.; et al. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Nitta, T.; Kuznetsov, Y.; McPherson, A.; Fan, H. Murine leukemia virus glycosylated Gag (gPr80gag) facilitates interferon-sensitive virus release through lipid rafts. Proc. Natl. Acad. Sci. USA 2010, 107, 1190–1195. [Google Scholar] [CrossRef]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef]
- Saad, J.S.; Ablan, S.D.; Ghanam, R.H.; Kim, A.; Andrews, K.; Nagashima, K.; Soheilian, F.; Freed, E.O.; Summers, M.F. Structure of the Myristylated Human Immunodeficiency Virus Type 2 Matrix Protein and the Role of Phosphatidylinositol-(4,5)-Bisphosphate in Membrane Targeting. J. Mol. Biol. 2008, 382, 434–447. [Google Scholar] [CrossRef]
- Sherer, N.M.; Lehmann, M.J.; Ingmundson, A.; Horner, S.M.; Cicchetti, G.; Allen, P.G.; Pypaert, M.; Cunningham, J.M.; Mothes, W.; Jimenez-Soto, L.F. Visualization of Retroviral Replication in Living Cells Reveals Budding into Multivesicular Bodies. Traffic 2003, 4, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Houzet, L.; Gay, B.; Morichaud, Z.; Briant, L.; Mougel, M. Intracellular assembly and budding of the Murine Leukemia Virus in infected cells. Retrovirology 2006, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Yeku, O.; Olepu, S.; Genna, A.; Park, J.-S.; Ren, H.; Du, G.; Gelb, M.H.; Morris, A.J.; Frohman, M.A. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a Phospholipase D Pharmacological Inhibitor That Alters Cell Spreading and Inhibits Chemotaxis. Mol. Pharmacol. 2009, 75, 437–446. [Google Scholar] [CrossRef]
- O’reilly, M.C.; Scott, S.A.; Brown, K.A.; Oguin, T.H.; Thomas, P.G.; Daniels, J.S.; Morrison, R.; Brown, H.A.; Lindsley, C.W. Development of Dual PLD1/2 and PLD2 Selective Inhibitors from a Common 1,3,8-Triazaspiro[4.5]decane Core: Discovery of ML298 and ML299 That Decrease Invasive Migration in U87-MG Glioblastoma Cells. J. Med. Chem. 2013, 56, 2695–2699. [Google Scholar] [CrossRef]
- Wright, B.D.; Loo, L.; Street, S.E.; Ma, A.; Taylor-Blake, B.; Stashko, M.A.; Jin, J.; Janzen, W.P.; Frye, S.V.; Zylka, M.J. The Lipid Kinase PIP5K1C Regulates Pain Signaling and Sensitization. Neuron 2014, 82, 836–847. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Yang, Y.-P.; Hu, L.-F.; Zheng, H.-F.; Mao, C.-J.; Hu, W.-D.; Xiong, K.-P.; Wang, F.; Liu, C.-F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Gao, G.; Luo, H. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 2006, 84, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Hamard-Peron, E.; Juillard, F.; Saad, J.S.; Roy, C.; Roingeard, P.; Summers, M.F.; Darlix, J.-L.; Picart, C.; Muriaux, D. Targeting of Murine Leukemia Virus Gag to the Plasma Membrane Is Mediated by PI(4,5)P2/PS and a Polybasic Region in the Matrix. J. Virol. 2010, 84, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Vitale, N.; Caumont, A.; Chasserot-Golaz, S.; Du, G.; Wu, S.; Sciorra, V.A.; Morris, A.J.; Frohman, M.A.; Bader, M. Phospholipase D1: A key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 2001, 20, 2424–2434. [Google Scholar] [CrossRef]
- Choi, W.S.; Kim, Y.M.; Combs, C.; Frohman, M.A.; Beaven, M.A. Phospholipases D1 and D2 Regulate Different Phases of Exocytosis in Mast Cells. J. Immunol. 2002, 168, 5682–5689. [Google Scholar] [CrossRef] [PubMed]
- Hughes, W.E.; Elgundi, Z.; Huang, P.; Frohman, M.A.; Biden, T.J. Phospholipase D1 regulates secretagogue-stimulated insulin release in pancreatic beta-cells. J. Biol. Chem. 2004, 279, 27534–27541. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-G.; Siddhanta, A.; Austin, C.D.; Hammond, S.M.; Sung, T.-C.; Frohman, M.A.; Morris, A.J.; Shields, D. Phospholipase D Stimulates Release of Nascent Secretory Vesicles from the trans-Golgi Network. J. Cell Biol. 1997, 138, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Roy, J.; Paquette, J.-S.; Fortin, J.-F.; Tremblay, M.J. The Immunosuppressant Rapamycin Represses Human Immunodeficiency Virus Type 1 Replication. Antimicrob. Agents Chemother. 2002, 46, 3447–3455. [Google Scholar] [CrossRef]
- Heredia, A.; Amoroso, A.; Davis, C.L.E.N.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Valiente-Echeverría, F.; Crossie, C.; Miron, M.-J.; Panté, N.; Olivier, M.; Mouland, A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Sci. Rep. 2017, 7, 5515. [Google Scholar] [CrossRef]
- Rose, N.J.; Lever, A.M. Rapamycin-induced inhibition of HTLV-I LTR activity is rescued by c-Myb. Retrovirology 2007, 4, 24. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Zhang, S.; Lin, X.; Hou, Q.; Hu, Z.; Wang, Y.; Wang, Z. Regulation of mTORC1 by amino acids in mammalian cells: A general picture of recent advances. Anim. Nutr. 2021, 7, 1009–1023. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Sowder, R.C.; Adams, J.; Schubert, U. Retroviruses Have Differing Requirements for Proteasome Function in the Budding Process. J. Virol. 2003, 77, 3384–3393. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Chau, V.; Wills, J.W. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 2000, 97, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the Vacuolar Protein Sorting Pathway Are Essential for HIV-1 Budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Segura-Morales, C.; Pescia, C.; Chatellard-Causse, C.; Sadoul, R.; Bertrand, E.; Basyuk, E. Tsg101 and Alix Interact with Murine Leukemia Virus Gag and Cooperate with Nedd4 Ubiquitin Ligases during Budding. J. Biol. Chem. 2005, 280, 27004–27012. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A.; Boross, P.; Sperka, T.; Miklóssy, G.; Kádas, J.; Bagossi, P.; Oroszlan, S.; Weber, I.T.; Tözsér, J. Characterization of the murine leukemia virus protease and its comparison with the human immunodeficiency virus type 1 protease. J. Gen. Virol. 2006, 87, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Diehl, N.; Schaal, H. Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [PubMed]
- Imam, S.; Talley, S.; Nelson, R.S.; Dharan, A.; O’Connor, C.; Hope, T.J.; Campbell, E.M. TRIM5α Degradation via Autophagy Is Not Required for Retroviral Restriction. J. Virol. 2016, 90, 3400–3410. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.R.; Schofield, J.J.; Farr, C.J.; Bucan, M. Mouse transferrin receptor 1 is the cell entry receptor for mouse mammary tumor virus. Proc. Natl. Acad. Sci. USA 2002, 99, 12386–12390. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Kang, T.; Jackson, L.; Murphy, A.; Nitta, T. Evidence for Involvement of ADP-Ribosylation Factor 6 in Intracellular Trafficking and Release of Murine Leukemia Virus Gag. Cells 2024, 13, 270. https://doi.org/10.3390/cells13030270
Kang H, Kang T, Jackson L, Murphy A, Nitta T. Evidence for Involvement of ADP-Ribosylation Factor 6 in Intracellular Trafficking and Release of Murine Leukemia Virus Gag. Cells. 2024; 13(3):270. https://doi.org/10.3390/cells13030270
Chicago/Turabian StyleKang, Hyokyun, Taekwon Kang, Lauryn Jackson, Amaiya Murphy, and Takayuki Nitta. 2024. "Evidence for Involvement of ADP-Ribosylation Factor 6 in Intracellular Trafficking and Release of Murine Leukemia Virus Gag" Cells 13, no. 3: 270. https://doi.org/10.3390/cells13030270