
SUMOylation Modulates Reactive Oxygen Species (ROS) Levels and Acts as a Protective Mechanism in the Type 2 Model of Diabetic Peripheral Neuropathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunoprecipitation and Western Blot Analysis
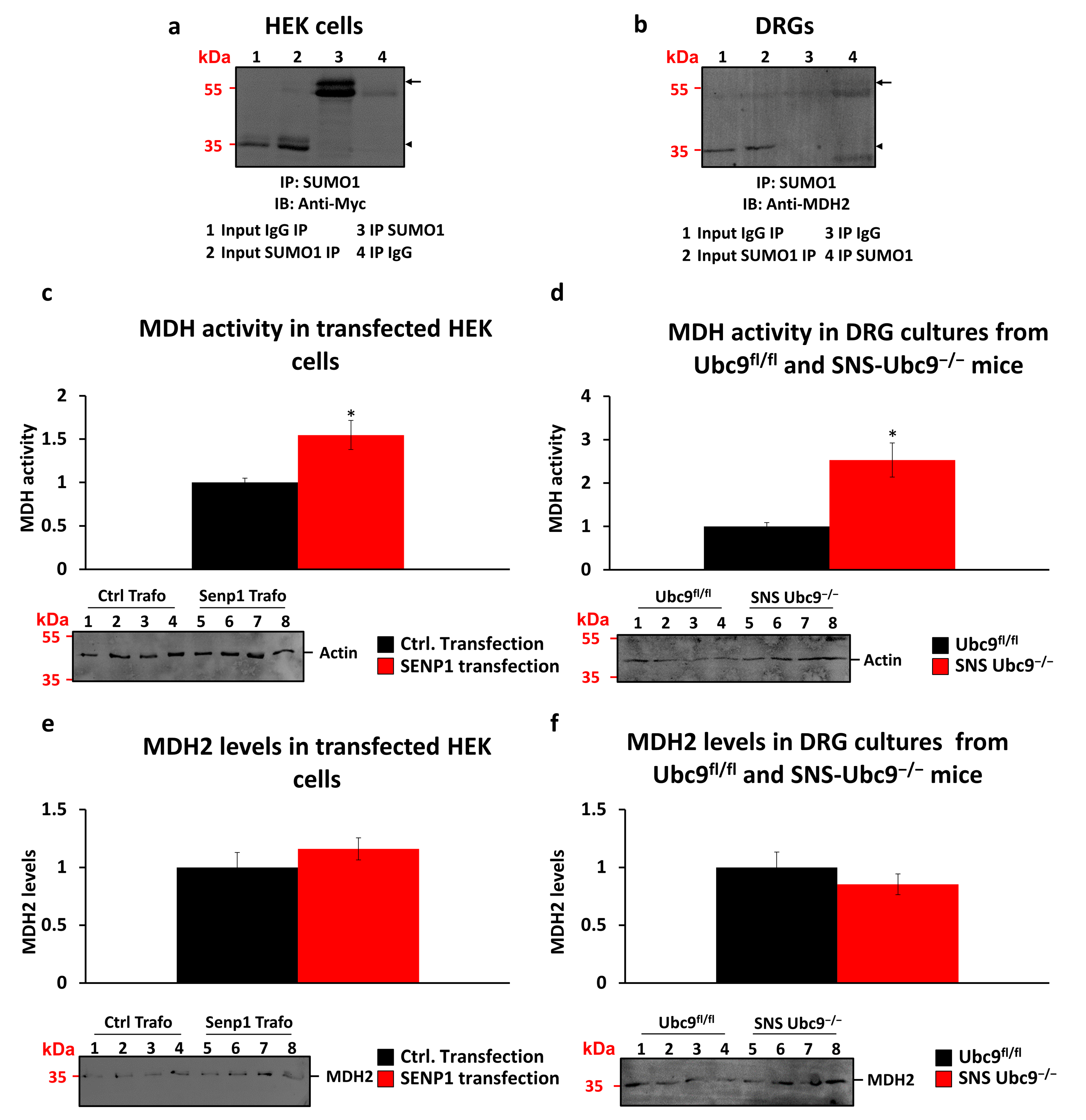
2.3. MDH Activity Assay
2.4. Animal Experiments
2.5. Sensory-Neuron-Specific Knockout Mice
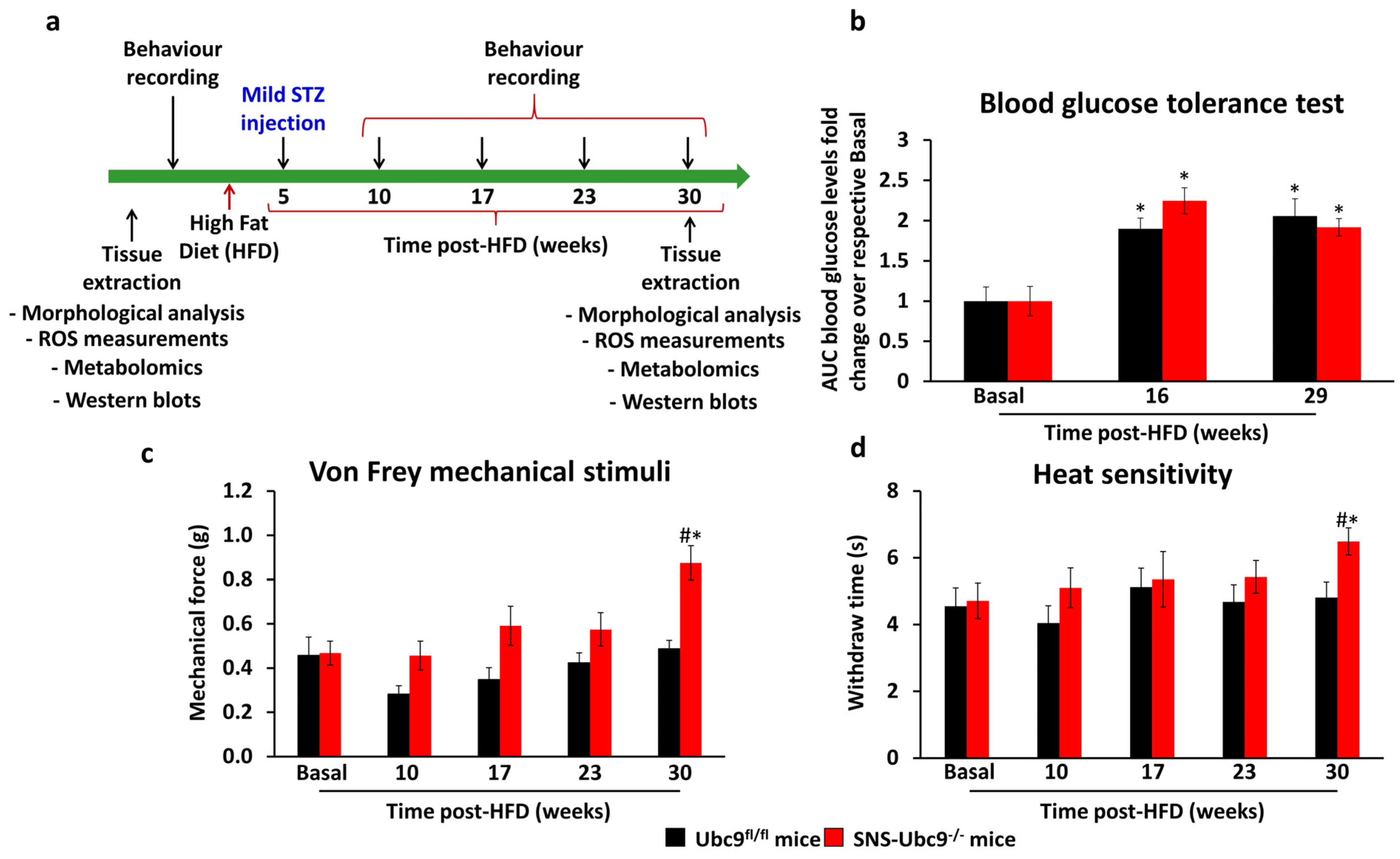
2.6. Rodent Model of Type 2 Diabetes
2.7. Behavioural Measurements
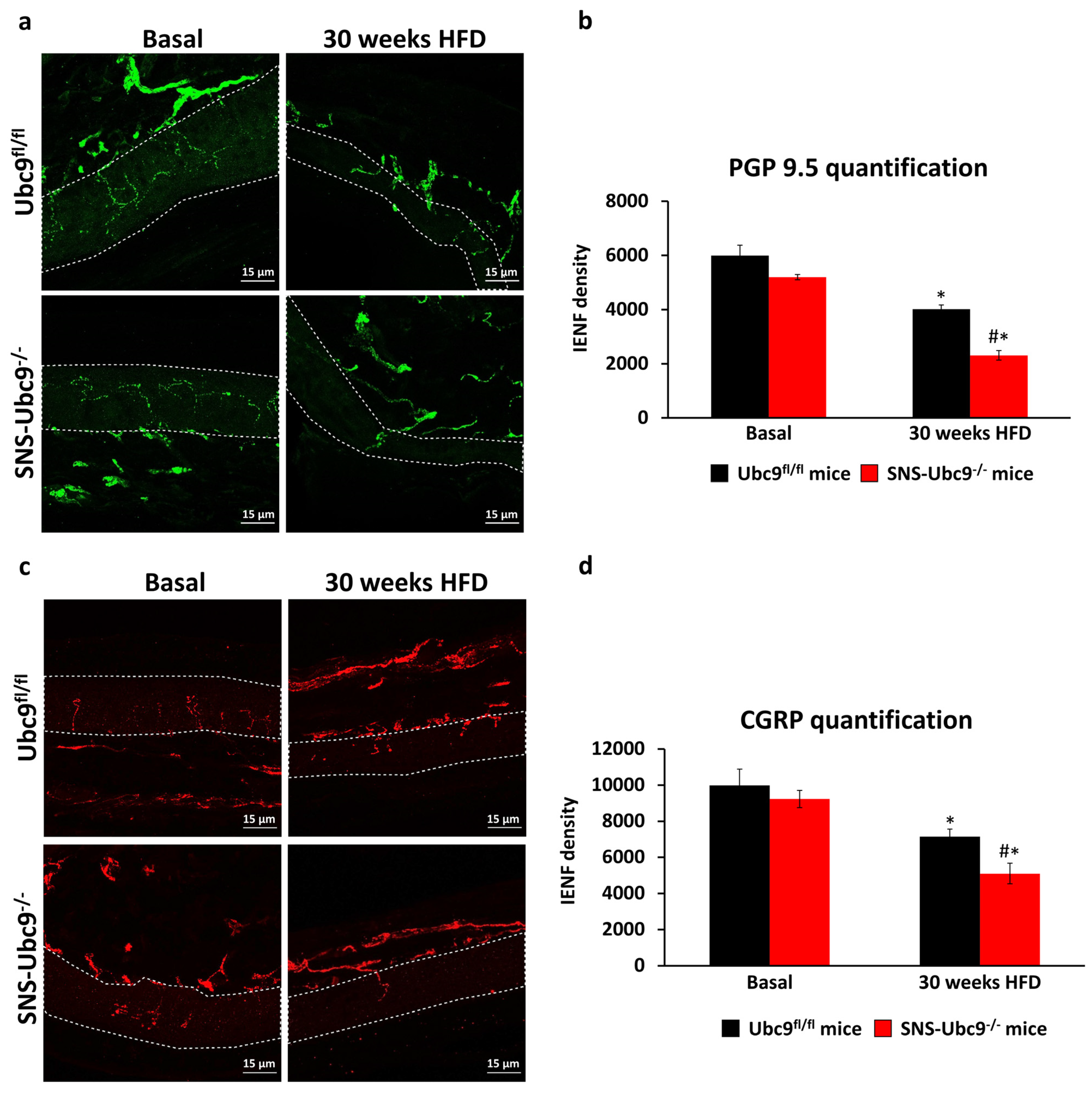
2.8. Immunohistochemistry
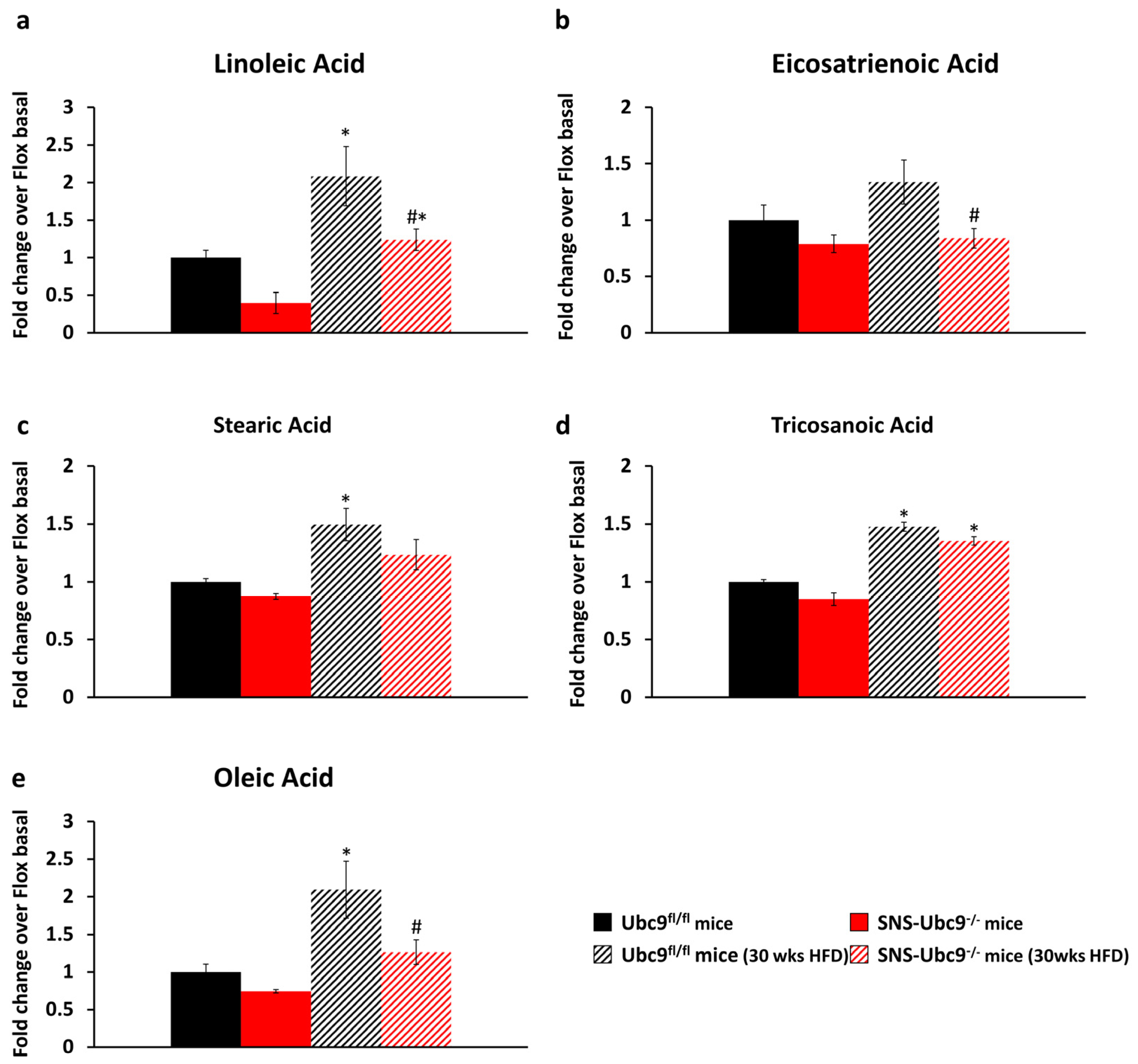
2.9. Metabolomics
2.9.1. Sample Extraction
2.9.2. Derivatisation (Methoximation and Silylation)
2.9.3. Gas Chromatography/Mass Spectrometry (GC/MS) Analysis
2.10. Statistics
3. Results
3.1. Mice Lacking SUMOylation, Specifically in DRG Sensory Neurons, Exhibit Accelerated DPN
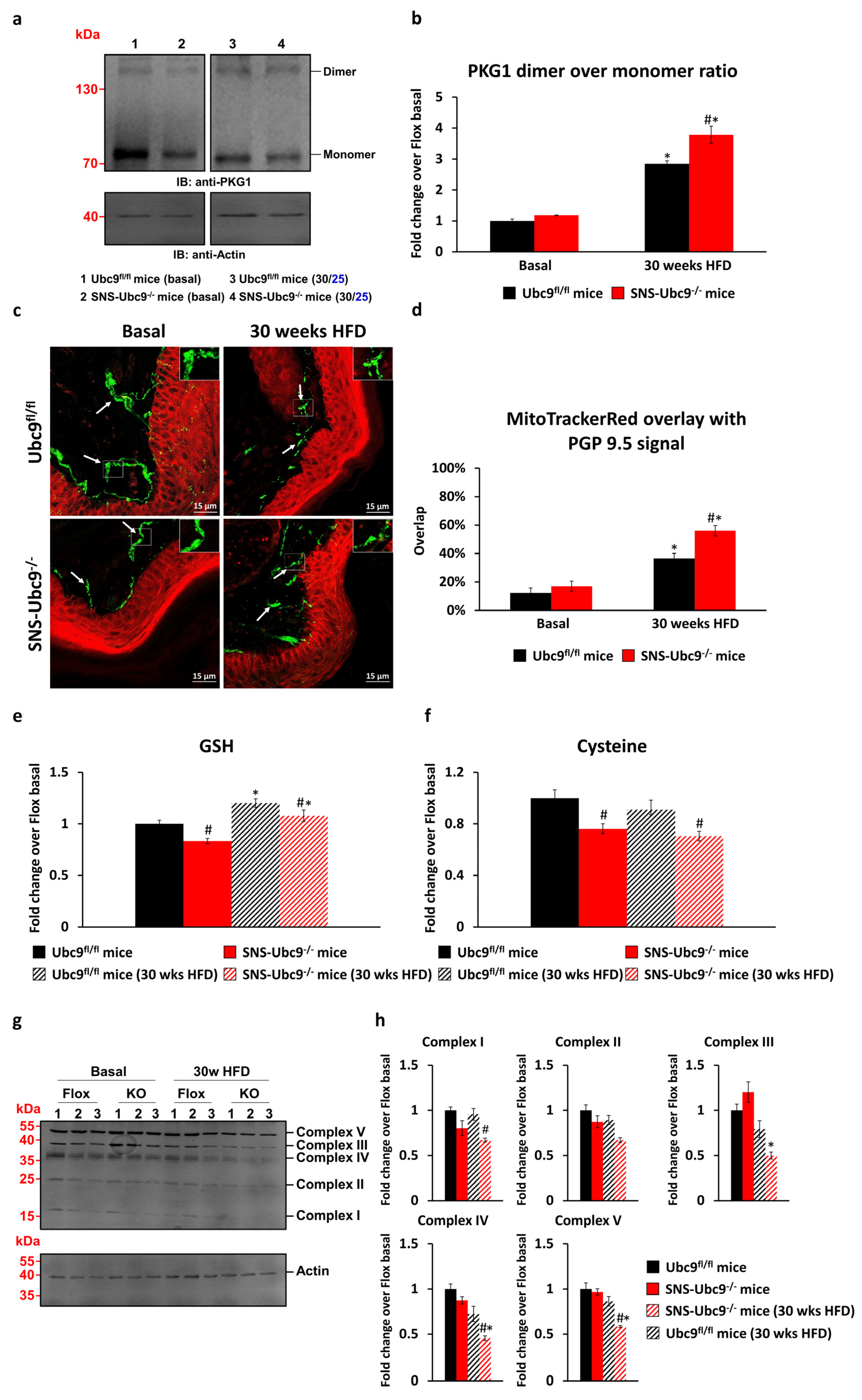
3.2. SUMOylation Modulates Reactive Oxygen Species (ROS) Levels in DRG Neurons and the Nerve Ending in Paw Skin of Mice with Type 2 Diabetes
3.3. Impact of SUMOylation on the Respiratory Chain
3.4. Malate Dehydrogenase as a SUMOylation Target
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, R.; Karuranga, S.; Malanda, B.; Saeedi, P.; Basit, A.; Besançon, S.; Bommer, C.; Esteghamati, A.; Ogurtsova, K.; Zhang, P.; et al. Global and regional estimates and projections of diabetes-related health expenditure: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2020, 162, 108072. [Google Scholar] [CrossRef]
- American Diabetes Association. Economic Costs of Diabetes in the U.S. in 2017. Diabetes Care 2018, 41, 917–928. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Reviews. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Allen, M.D.; Choi, I.H.; Kimpinski, K.; Doherty, T.J.; Rice, C.L. Motor unit loss and weakness in association with diabetic neuropathy in humans. Muscle Nerve 2013, 48, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Zakin, E.; Abrams, R.; Simpson, D.M. Diabetic Neuropathy. Semin. Neurol. 2019, 39, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.C.; Cheng, H.T.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef]
- Jensen, T.S.; Karlsson, P.; Gylfadottir, S.S.; Andersen, S.T.; Bennett, D.L.; Tankisi, H.; Finnerup, N.B.; Terkelsen, A.J.; Khan, K.; Themistocleous, A.C.; et al. Painful and non-painful diabetic neuropathy, diagnostic challenges and implications for future management. Brain A J. Neurol. 2021, 144, 1632–1645. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, D.; Kar, D.; Khunti, K.; Davies, M.J.; Scott, A.R.; Walker, J.; Tesfaye, S. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019, 7, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef]
- Kobayashi, M.; Zochodne, D.W. Diabetic neuropathy and the sensory neuron: New aspects of pathogenesis and their treatment implications. J. Diabetes Investig. 2018, 9, 1239–1254. [Google Scholar] [CrossRef]
- McLaughlin, R.J.; Spindler, M.P.; van Lummel, M.; Roep, B.O. Where, How, and When: Positioning Posttranslational Modification within Type 1 Diabetes Pathogenesis. Curr. Diabetes Rep. 2016, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Al-Saoudi, E.; Christensen, M.M.B.; Nawroth, P.; Fleming, T.; Hommel, E.E.; Jørgensen, M.E.; Fleischer, J.; Hansen, C.S. Advanced glycation end-products are associated with diabetic neuropathy in young adults with type 1 diabetes. Front. Endocrinol. 2022, 13, 891442. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Peruzzotti-Jametti, L.; Leonardi, T.; Vicario, N.; Ye, D.; Lee, Y.-J.; Maric, D.; Johnson, K.R.; Mou, Y.; Bosch, A.V.D.; et al. SUMOylation promotes survival and integration of neural stem cell grafts in ischemic stroke. EBioMedicine 2019, 42, 214–224. [Google Scholar] [CrossRef]
- Hoppe, J.B.; Salbego, C.G.; Cimarosti, H. SUMOylation: Novel Neuroprotective Approach for Alzheimer’s Disease? Aging Dis. 2015, 6, 322–330. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Yang, W.; Ye, D.G.; Shen, Y.; Pluchino, S.; Lee, Y.-J.; Hallenbeck, J.M.; Paschen, W. SUMOylation in brain ischemia: Patterns, targets, and translational implications. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 5–16. [Google Scholar] [CrossRef]
- Agarwal, N.; Taberner, F.J.; Rojas, D.R.; Moroni, M.; Omberbasic, D.; Njoo, C.; Andrieux, A.; Gupta, P.; Bali, K.K.; Herpel, E.; et al. SUMOylation of Enzymes and Ion Channels in Sensory Neurons Protects against Metabolic Dysfunction, Neuropathy, and Sensory Loss in Diabetes. Neuron 2020, 107, 1141–1159.e7. [Google Scholar] [CrossRef]
- Hou, Z.; Chen, J.; Yang, H.; Hu, X.; Yang, F. PIAS1 alleviates diabetic peripheral neuropathy through SUMOlation of PPAR-γ and miR-124-induced downregulation of EZH2/STAT3. Cell Death Discov. 2021, 7, 372. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Rajan, S.; Plant, L.D.; Rabin, M.L.; Butler, M.H.; Goldstein, S.A.N. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 2005, 121, 37–47. [Google Scholar] [CrossRef]
- Xiong, D.; Li, T.; Dai, H.; Arena, A.F.; Plant, L.D.; Goldstein, S.A.N. SUMOylation determines the voltage required to activate cardiac I Ks channels. Proc. Natl. Acad. Sci. USA 2017, 114, E6686–E6694. [Google Scholar] [CrossRef]
- Um, J.W.; Chung, K.C. Functional modulation of parkin through physical interaction with SUMO-1. J. Neurosci. Res. 2006, 84, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Villegas-Rodríguez, M.E.; Uribarri, J.; Solorio-Meza, S.E.; Fajardo-Araujo, M.E.; Cai, W.; Torres-Graciano, S.; Rangel-Salazar, R.; Wrobel, K.; Garay-Sevilla, M.E. The AGE-RAGE Axis and Its Relationship to Markers of Cardiovascular Disease in Newly Diagnosed Diabetic Patients. PLoS ONE 2016, 11, e0159175. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, L.; Yang, R.; Feng, R.; Li, Z.; Zhou, X.; Dong, Z.; Ghartey-Kwansah, G.; Xu, M.; Nishi, M.; et al. Optimizing conditions for calcium phosphate mediated transient transfection. Saudi J. Biol. Sci. 2017, 24, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, M.; Agarwal, N.; Stösser, S.; Bali, K.K.; Karaulanov, E.; Kamble, R.; Pospisilova, B.; Kurejova, M.; Birchmeier, W.; Niehrs, C.; et al. Wnt-Fzd Signaling Sensitizes Peripheral Sensory Neurons via Distinct Noncanonical Pathways. Neuron 2014, 83, 104–121. [Google Scholar] [CrossRef]
- Becker, J.; Barysch, S.V.; Karaca, S.; Dittner, C.; Hsiao, H.-H.; Berriel Diaz, M.; Herzig, S.; Urlaub, H.; Melchior, F. Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol. 2013, 20, 525–531. [Google Scholar] [CrossRef]
- Valek, L.; Häussler, A.; Dröse, S.; Eaton, P.; Schröder, K.; Tegeder, I. Redox-guided axonal regrowth requires cyclic GMP dependent protein kinase 1: Implication for neuropathic pain. Redox Biol. 2017, 11, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Pillai-Kastoori, L.; Schutz-Geschwender, A.R.; Harford, J.A. A systematic approach to quantitative Western blot analysis. Anal. Biochem. 2020, 593, 113608. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.S.; Blobel, G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J. Biol. Chem. 1997, 272, 26799–26802. [Google Scholar] [CrossRef]
- Seufert, W.; Futcher, B.; Jentsch, S. Role of a ubiquitin-conjugating enzyme in degradation of S- and M-phase cyclins. Nature 1995, 373, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, W.M.; Lountos, G.T.; Zlotkowski, K.; Dahlhauser, S.D.; Saunders, L.B.; Needle, D.; Tropea, J.E.; Zhan, C.; Wei, G.; Ma, B.; et al. Insights Into the Allosteric Inhibition of the SUMO E2 Enzyme Ubc9. Angew. Chem. Int. Ed. 2016, 55, 5703–5707. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Offermanns, S.; Kuner, R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis 2004, 38, 122–129. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Sakowski, S.A.; Feldman, E.L. Mouse models of diabetic neuropathy. ILAR J. 2014, 54, 259–272. [Google Scholar] [CrossRef]
- Gilbert, E.R.; Fu, Z.; Liu, D. Development of a nongenetic mouse model of type 2 diabetes. Exp. Diabetes Res. 2011, 2011, 416254. [Google Scholar] [CrossRef]
- Agarwal, N.; Pacher, P.; Tegeder, I.; Amaya, F.; E Constantin, C.; Brenner, G.J.; Rubino, T.; Michalski, C.W.; Marsicano, G.; Monory, K.; et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat. Neurosci. 2007, 10, 870–879. [Google Scholar] [CrossRef]
- Tappe, A.; Klugmann, M.; Luo, C.; Hirlinger, D.; Agarwal, N.; Benrath, J.; Ehrengruber, M.U.; During, M.J.; Kuner, R. Synaptic scaffolding protein Homer1a protects against chronic inflammatory pain. Nat. Med. 2006, 12, 677–681. [Google Scholar] [CrossRef]
- Rojas, D.R.; Kuner, R.; Agarwal, N. Metabolomic signature of type 1 diabetes-induced sensory loss and nerve damage in diabetic neuropathy. J. Mol. Med. 2019, 97, 845–854. [Google Scholar] [CrossRef]
- Rojas, D.R.; Tegeder, I.; Kuner, R.; Agarwal, N. Hypoxia-inducible factor 1α protects peripheral sensory neurons from diabetic peripheral neuropathy by suppressing accumulation of reactive oxygen species. J. Mol. Med. 2018, 96, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan, V.; Zheng, H.; Taberner, F.J.; Landry, J.; Nees, T.A.; Pistolic, J.; Agarwal, N.; Männich, D.; Benes, V.; Helmstaedter, M.; et al. Neuropathic pain caused by miswiring and abnormal end organ targeting. Nature 2022, 606, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Gangadharan, V.; Liu, S.; Körber, C.; Tan, L.L.; Li, H.; Oswald, M.J.; Kang, J.; Martin-Cortecero, J.; Männich, D.; et al. Layer-specific pain relief pathways originating from primary motor cortex. Science 2022, 378, 1336–1343. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.-S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef]
- Santa-María, C.; López-Enríquez, S.; la Paz, S.M.-D.; Geniz, I.; Reyes-Quiroz, M.E.; Moreno, M.; Palomares, F.; Sobrino, F.; Alba, G. Update on Anti-Inflammatory Molecular Mechanisms Induced by Oleic Acid. Nutrients 2023, 15, 224. [Google Scholar] [CrossRef]
- Mandel, N.; Agarwal, N. Role of SUMOylation in Neurodegenerative Diseases. Cells 2022, 11, 3395. [Google Scholar] [CrossRef]
- Soares, E.S.; Prediger, R.D.; Brocardo, P.S.; Cimarosti, H.I. SUMO-modifying Huntington’s disease. IBRO Neurosci. Rep. 2022, 12, 203–209. [Google Scholar] [CrossRef]
- Kamynina, E.; Stover, P.J. The Roles of SUMO in Metabolic Regulation. Adv. Exp. Med. Biol. 2017, 963, 143–168. [Google Scholar] [CrossRef]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Camacho, L.R.; Kormanovski, A.; Del Carmen Castillo-Hernández, M.; Guevara-Balcázar, G.; Lara-Padilla, E. Alterations in glutathione, nitric oxide and 3-nitrotyrosine levels following exercise and/or hyperbaric oxygen treatment in mice with diet-induced diabetes. Biomed. Rep. 2020, 12, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Trovato, A.E.; Comelli, F.; Giagnoni, G.; Colleoni, M. The non-psychoactive cannabis constituent cannabidiol is an orally effective therapeutic agent in rat chronic inflammatory and neuropathic pain. Eur. J. Pharmacol. 2007, 556, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Guedes, R.P.; Bosco, L.D.; Da Araújo, A.S.R.; Belló-Klein, A.; Ribeiro, M.F.M.; Partata, W.A. Sciatic nerve transection increases gluthatione antioxidant system activity and neuronal nitric oxide synthase expression in the spinal cord. Brain Res. Bull. 2009, 80, 422–427. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Reichart, G.; Mayer, J.; Zehm, C.; Kirschstein, T.; Tokay, T.; Lange, F.; Baltrusch, S.; Tiedge, M.; Fuellen, G.; Ibrahim, S.; et al. Mitochondrial complex IV mutation increases reactive oxygen species production and reduces lifespan in aged mice. Acta Physiol. 2019, 225, e13214. [Google Scholar] [CrossRef]
- Walters, T.S.; McIntosh, D.J.; Ingram, S.M.; Tillery, L.; Motley, E.D.; Arinze, I.J.; Misra, S. SUMO-Modification of Human Nrf2 at K110 and K533 Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell. Physiol. Biochem. 2021, 55, 141–159. [Google Scholar] [CrossRef]
- He, J.; Cheng, J.; Wang, T. SUMOylation-Mediated Response to Mitochondrial Stress. Int. J. Mol. Sci. 2020, 21, 5657. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Bourboulia, D.; Beebe, K.; Woodford, M.R.; Polier, S.; Hoang, A.; Chelluri, R.; Li, Y.; Guo, A.; Lee, M.-J.; et al. Asymmetric Hsp90 N Domain SUMOylation Recruits Aha1 and ATP-Competitive Inhibitors. Mol. Cell 2014, 53, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.-J. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. Targets Ther. 2016, 9, 145–153. [Google Scholar] [CrossRef]
- Kwon, H.J.; Hahn, K.R.; Kang, M.S.; Choi, J.H.; Moon, S.M.; Yoon, Y.S.; Hwang, I.K.; Kim, D.W. Tat-malate dehydrogenase fusion protein protects neurons from oxidative and ischemic damage by reduction of reactive oxygen species and modulation of glutathione redox system. Sci. Rep. 2023, 13, 5653. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G.S.; Wang, L.; Schroder, E.A.; Callahan, L.A.P. MitoTEMPOL, a mitochondrial targeted antioxidant, prevents sepsis-induced diaphragm dysfunction. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L228–L238. [Google Scholar] [CrossRef]
- Ban, H.S.; Xu, X.; Jang, K.; Kim, I.; Kim, B.-K.; Lee, K.; Won, M. A Novel Malate Dehydrogenase 2 Inhibitor Suppresses Hypoxia-Inducible Factor-1 by Regulating Mitochondrial Respiration. PLoS ONE 2016, 11, e0162568. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Malate dehydrogenase-2 inhibitor LW6 promotes metabolic adaptations and reduces proliferation and apoptosis in activated human T-cells. Exp. Ther. Med. 2015, 10, 1959–1966. [Google Scholar] [CrossRef]
- Krajnak, K.; Dahl, R. Small molecule SUMOylation activators are novel neuroprotective agents. Bioorganic Med. Chem. Lett. 2018, 28, 405–409. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef]
- Kumar, A.; Ito, A.; Takemoto, M.; Yoshida, M.; Zhang, K.Y.J. Identification of,5-oxadiazoles as a new class of SENP2 inhibitors using structure based virtual screening. J. Chem. Inf. Model. 2014, 54, 870–880. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; E Deverman, B.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandel, N.; Büttner, M.; Poschet, G.; Kuner, R.; Agarwal, N. SUMOylation Modulates Reactive Oxygen Species (ROS) Levels and Acts as a Protective Mechanism in the Type 2 Model of Diabetic Peripheral Neuropathy. Cells 2023, 12, 2511. https://doi.org/10.3390/cells12212511
Mandel N, Büttner M, Poschet G, Kuner R, Agarwal N. SUMOylation Modulates Reactive Oxygen Species (ROS) Levels and Acts as a Protective Mechanism in the Type 2 Model of Diabetic Peripheral Neuropathy. Cells. 2023; 12(21):2511. https://doi.org/10.3390/cells12212511
Chicago/Turabian StyleMandel, Nicolas, Michael Büttner, Gernot Poschet, Rohini Kuner, and Nitin Agarwal. 2023. "SUMOylation Modulates Reactive Oxygen Species (ROS) Levels and Acts as a Protective Mechanism in the Type 2 Model of Diabetic Peripheral Neuropathy" Cells 12, no. 21: 2511. https://doi.org/10.3390/cells12212511