
The Role of Polo-Like Kinase 1 in Regulating the Forkhead Box Family Transcription Factors

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
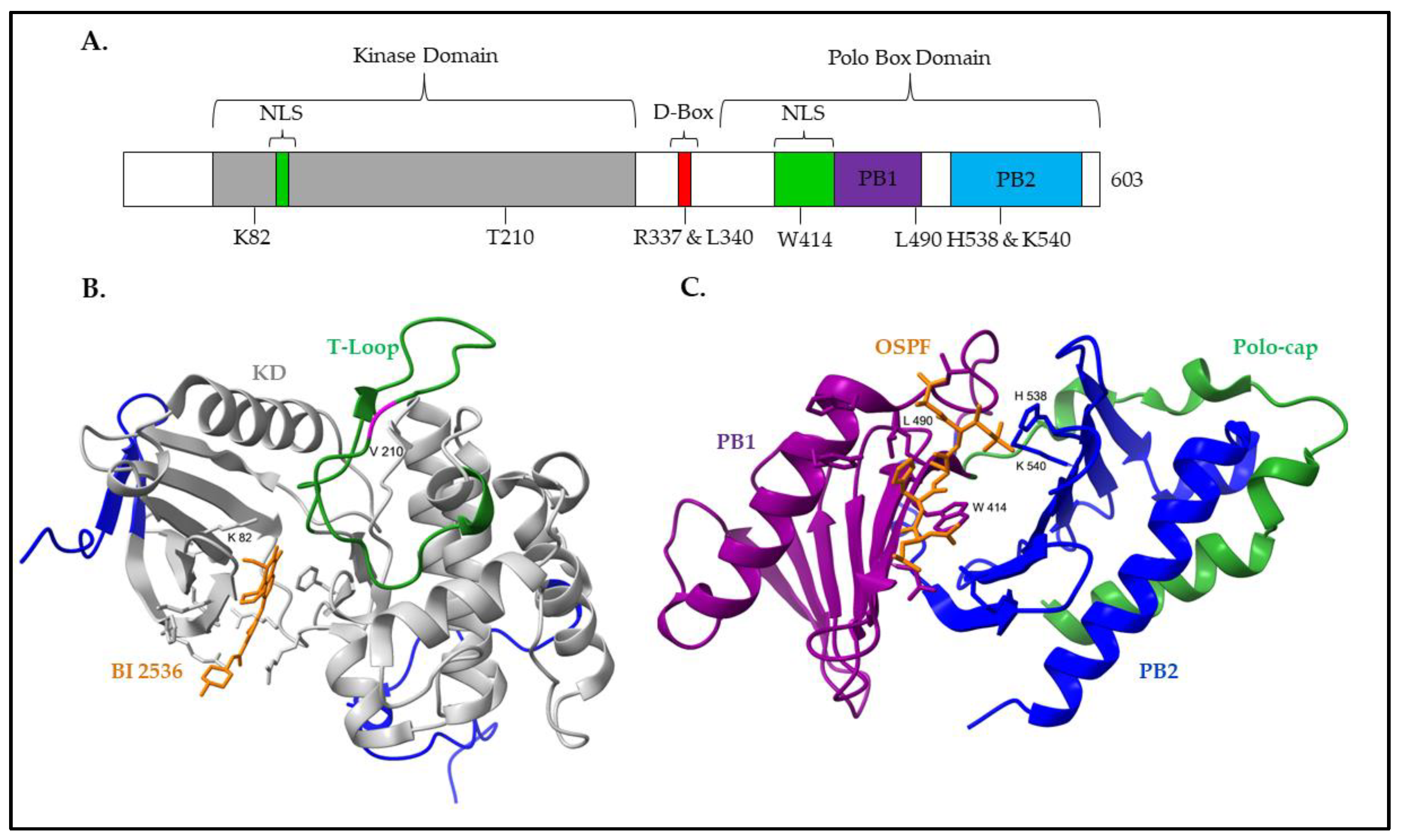
2. Structure and Regulation of PLK1
2.1. Structure of PLK1
2.2. Regulation of PLK1
2.2.1. Regulation of PLK1 Expression
2.2.2. Regulation of PLK1 Stability
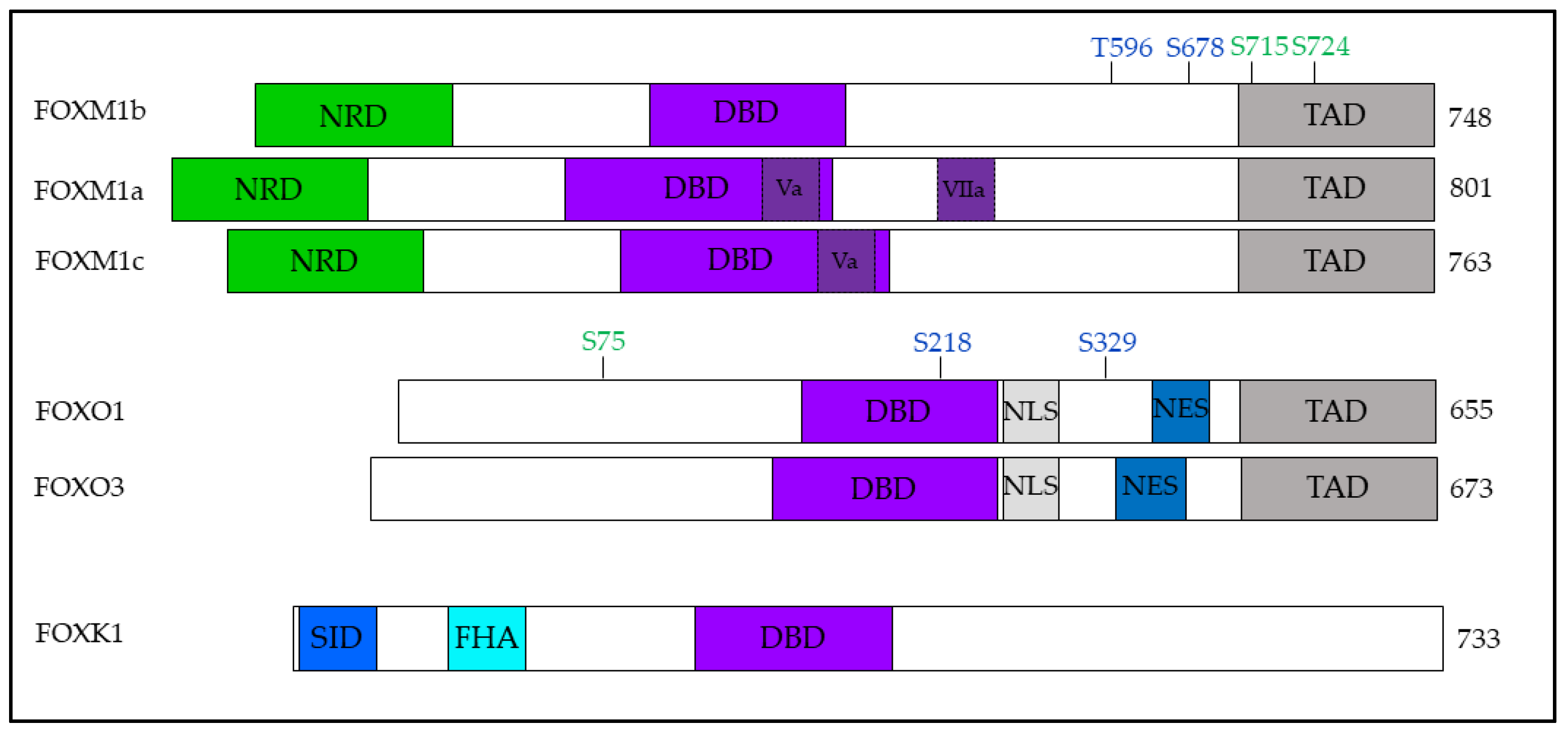
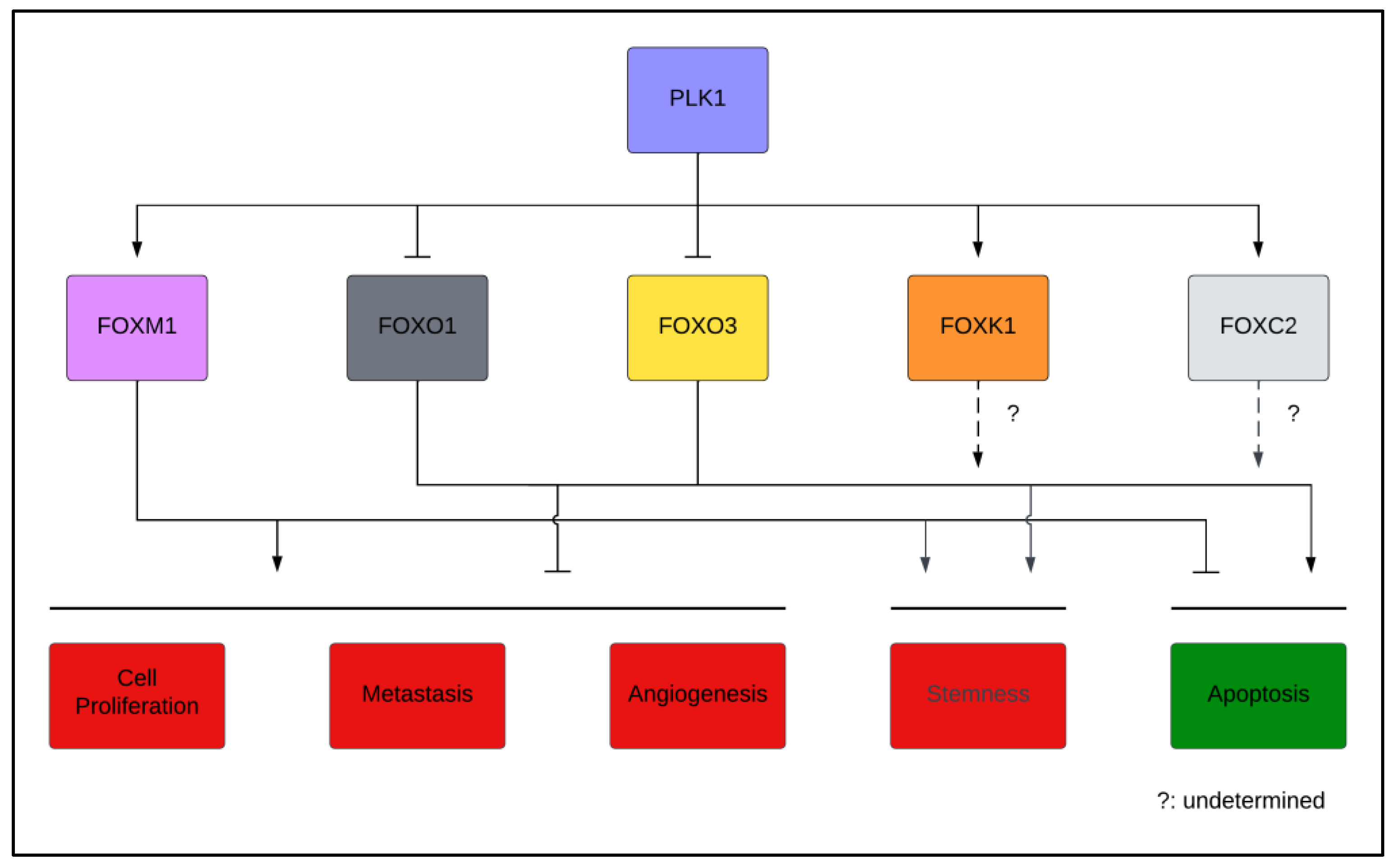
3. Regulation of the FOX Family by PLK1
3.1. FOXM1
3.1.1. Background
3.1.2. Functions
3.1.3. Regulation
3.2. FOXO1 and FOXO3
3.2.1. FOXO1 and FOXO3 Background
3.2.2. FOXO1 and FOXO3 Functions
3.2.3. FOXO1 and FOXO3 Regulation
3.3. FOXK1
3.3.1. Background
3.3.2. Functions
3.3.3. Regulation
3.4. Other FOX Transcription Factors
4. The Clinical Significance of PLK1 and FOX Transcription Factors
4.1. PLK1 and FOX Transcription Factors in Cancer
4.2. PLK1 Inhibition
4.3. Disruption of PLK1-Regulated FOX Transcription Factor Signaling
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chase, D.; Golden, A.; Heidecker, G.; Ferris, D.K. Caenorhabditis Elegans Contains a Third Polo-like Kinase Gene. DNA Seq. 2000, 11, 327–334. [Google Scholar] [CrossRef]
- Andrysik, Z.; Bernstein, W.Z.; Deng, L.; Myer, D.L.; Li, Y.-Q.; Tischfield, J.A.; Stambrook, P.J.; Bahassi, E.M. The Novel Mouse Polo-like Kinase 5 Responds to DNA Damage and Localizes in the Nucleolus. Nucleic Acids Res. 2010, 38, 2931–2943. [Google Scholar] [CrossRef] [PubMed]
- Sunkel, C.E.; Glover, D.M. Polo, a Mitotic Mutant of Drosophila Displaying Abnormal Spindle Poles. J. Cell Sci. 1988, 89, 25–38. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional Evolution of Polo-like Kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.-Y.; Wood, J.L.; Minter-Dykhouse, K.; Ye, L.; Saunders, T.L.; Yu, X.; Chen, J. Polo-Like Kinase 1 Is Essential for Early Embryonic Development and Tumor Suppression. Mol. Cell. Biol. 2008, 28, 6870–6876. [Google Scholar] [CrossRef]
- Petronczki, M.; Lénárt, P.; Peters, J.M. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev. Cell 2008, 14, 646–659. [Google Scholar] [CrossRef]
- Schmucker, S.; Sumara, I. Molecular Dynamics of PLK1 during Mitosis. Mol. Cell. Oncol. 2014, 1, e954507. [Google Scholar] [CrossRef]
- Raab, C.A.; Raab, M.; Becker, S.; Strebhardt, K. Non-Mitotic Functions of Polo-like Kinases in Cancer Cells. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188467. [Google Scholar] [CrossRef]
- Kressin, M.; Fietz, D.; Becker, S.; Strebhardt, K. Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer. Cells 2021, 10, 1176. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 2021, 13, 1011. [Google Scholar] [CrossRef] [PubMed]
- Lowery, D.M.; Clauser, K.R.; Hjerrild, M.; Lim, D.; Alexander, J.; Kishi, K.; Ong, S.E.; Gammeltoft, S.; Carr, S.A.; Yaffe, M.B. Proteomic Screen Defines the Polo-Box Domain Interactome and Identifies Rock2 as a Plk1 Substrate. EMBO J. 2007, 26, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like Kinase 1 (PLK1) Signaling in Cancer and Beyond. Biochem. Pharmacol. 2021, 193, 114747. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.E.; Macauley, M.J.; Vizeacoumar, F.S.; Abuhussein, O.; Freywald, A.; Vizeacoumar, F.J. The CINs of Polo-Like Kinase 1 in Cancer. Cancers 2020, 12, 2953. [Google Scholar] [CrossRef] [PubMed]
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016. [Google Scholar] [CrossRef] [PubMed]
- Gheghiani, L.; Wang, L.; Zhang, Y.; Moore, X.T.R.; Zhang, J.; Smith, S.C.; Tian, Y.; Wang, L.; Turner, K.; Jackson-Cook, C.K.; et al. PLK1 Induces Chromosomal Instability and Overrides Cell-Cycle Checkpoints to Drive Tumorigenesis. Cancer Res. 2021, 81, 1293–1307. [Google Scholar] [CrossRef]
- Karlin, K.L.; Mondal, G.; Hartman, J.K.; Tyagi, S.; Kurley, S.J.; Bland, C.S.; Hsu, T.Y.T.; Renwick, A.; Fang, J.E.; Migliaccio, I.; et al. The Oncogenic STP Axis Promotes Triple-Negative Breast Cancer via Degradation of the REST Tumor Suppressor. Cell Rep. 2014, 9, 1318–1332. [Google Scholar] [CrossRef]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like Kinase 1 (Plk1) Inhibits P53 Function by Physical Interaction and Phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-Dependent Phosphorylation of FoxM1 Regulates a Transcriptional Programme Required for Mitotic Progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef]
- Yuan, C.; Wang, L.; Zhou, L.; Fu, Z. The Function of FOXO1 in the Late Phases of the Cell Cycle Is Suppressed by PLK1-Mediated Phosphorylation. Cell Cycle 2014, 13, 807–819. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Rennie, G.R.; Feru, F.; Kuhn, C.; Ding, Y.-H. Selectivity-Determining Residues in Plk1. Chem. Biol. Drug Des. 2007, 70, 540–546. [Google Scholar] [CrossRef]
- Ding, Y.-H.; Kothe, M.; Kohls, D.; Low, S. Structure of PLK1 in Complex with BI2536. 2008. Available online: https://doi.org/10.2210/pdb2RKU/pdb (accessed on 4 March 2023).
- Sharma, P.; Mahen, R.; Rossmann, M.; Stokes, J.E.; Hardwick, B.; Huggins, D.J.; Emery, A.; Kunciw, D.L.; Hyvönen, M.; Spring, D.R.; et al. A Cryptic Hydrophobic Pocket in the Polo-Box Domain of the Polo-like Kinase PLK1 Regulates Substrate Recognition and Mitotic Chromosome Segregation. Sci. Rep. 2019, 9, 15930. [Google Scholar] [CrossRef]
- Kunciw, D.L.; Rossmann, M.; De Fusco, C.; Spring, D.R.; Hyvonen, M. The Structure of the Polo-Box Domain (PBD) of Polo-like Kinase 1 (Plk1) in Complex with LHSpTA Peptide. 2017. Available online: https://doi.org/10.2210/pdb5NFU/pdb (accessed on 4 March 2023).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The Protein Kinase Family: Conserved Features and Deduced Phylogeny of the Catalytic Domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Clay, F.J.; McEwen, S.J.; Bertoncello, I.; Wilks, A.F.; Dunn, A.R. Identification and Cloning of a Protein Kinase-Encoding Mouse Gene, Plk, Related to the Polo Gene of Drosophila. Proc. Natl. Acad. Sci. USA 1993, 90, 4882–4886. [Google Scholar] [CrossRef]
- Nakojima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a Consensus Motif for PlK (Polo-like Kinase) Phosphorylation Reveals Myt1 as a Plk1 Substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative Phosphoproteomics Identifies Substrates and Functional Modules of Aurora and Polo-like Kinase Activities in Mitotic Cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [PubMed]
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Cheng, A.C.; Jacques, S.L.; Johnson, T.L.; Lewis, C.; Loh, C.; Nonomiya, J.; et al. Structure of the Catalytic Domain of Human Polo-like Kinase 1. Biochemistry 2007, 46, 5960–5971. [Google Scholar] [CrossRef]
- McInnes, C.; Mezna, M.; Fischer, P. Progress in the Discovery of Polo-like Kinase Inhibitors. Curr. Top. Med. Chem. 2005, 5, 181–197. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krššák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.F.; Stewart, K.D.; Woods, K.W.; Giranda, V.L.; Luo, Y. Pharmacological and Functional Comparison of the Polo-like Kinase Family: Insight into Inhibitor and Substrate Specificity. Biochemistry 2007, 46, 9551–9563. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-Y.; Lowe, E.D.; Sinclair, J.; Nigg, E.A.; Johnson, L.N. The Crystal Structure of the Human Polo-like Kinase-1 Polo Box Domain and Its Phospho-Peptide Complex. Eur. Mol. Biol. Org. J. 2003, 22, 5757–5768. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.H.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The Molecular Basis for Phosphodependent Substrate Targeting and Regulation of Plks by the Polo-Box Domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Cantley, L.C.; Yaffe, M.B. Proteomic Screen Finds PSer/PThr-Binding Domain Localizing Plk1 to Mitotic Substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef]
- Park, J.-E.; Soung, N.-K.; Johmura, Y.; Kang, Y.H.; Liao, C.; Lee, K.H.; Park, C.H.; Nicklaus, M.C.; Lee, K.S. Polo-Box Domain: A Versatile Mediator of Polo-like Kinase Function. Cell. Mol. Life Sci. 2010, 67, 1957–1970. [Google Scholar] [CrossRef]
- Neef, R.; Gruneberg, U.; Kopajtich, R.; Li, X.; Nigg, E.A.; Sillje, H.; Barr, F.A. Choice of Plk1 Docking Partners during Mitosis and Cytokinesis Is Controlled by the Activation State of Cdk1. Nat. Cell Biol. 2007, 9, 436–444. [Google Scholar] [CrossRef]
- Archambault, V.; D’Avino, P.P.; Deery, M.J.; Lilley, K.S.; Glover, D.M. Sequestration of Polo Kinase to Microtubules by Phosphopriming-Independent Binding to Map205 Is Relieved by Phosphorylation at a CDK Site in Mitosis. Genes Dev. 2008, 22, 2707–2720. [Google Scholar] [CrossRef]
- Leung, G.C.; Hudson, J.W.; Kozarova, A.; Davidson, A.; Dennis, J.W.; Sicheri, F. The Sak Polo-Box Comprises a Structural Domain Sufficient for Mitotic Subcellular Localization. Nat. Struct. Biol. 2002, 9, 719–724. [Google Scholar] [CrossRef]
- Pintard, L.; Archambault, V. A Unified View of Spatio-Temporal Control of Mitotic Entry: Polo Kinase as the Key. Open Biol. 2018, 8, 180114. [Google Scholar] [CrossRef]
- Anger, M.; Kues, W.A.; Klima, J.; Mielenz, M.; Kubelka, M.; Motlik, J.; Esner, M.; Dvorak, P.; Carnwath, J.W.; Niemann, H. Cell Cycle Dependent Expression of Plk1 in Synchronized Porcine Fetal Fibroblasts. Mol. Reprod. Dev. 2003, 65, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Uchiumi, T.; Longo, D.L.; Ferris, D.K. Cell Cycle Regulation of the Human Polo-like Kinase (PLK) Promoter. J. Biol. Chem. 1997, 272, 9166–9174. [Google Scholar] [CrossRef] [PubMed]
- Nettelbeck, D.M.; Jérôme, V.; Müller, R. A Dual Specificity Promoter System Combining Cell Cycle-Regulated and Tissue-Specific Transcriptional Control. Gene Ther. 1999, 6, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Tategu, M.; Nakagawa, H.; Sasaki, K.; Yamauchi, R.; Sekimachi, S.; Suita, Y.; Watanabe, N.; Yoshid, K. Transcriptional Regulation of Human Polo-like Kinases and Early Mitotic Inhibitor. J. Genet. Genom. 2008, 35, 215–224. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 Is Required for Execution of the Mitotic Programme and Chromosome Stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Chen, X.; Muller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The Forkhead Transcription Factor FOXM1 Controls Cell Cycle-Dependent Gene Expression through an Atypical Chromatin Binding Mechanism. Mol. Cell. Biol. 2013, 33, 227–236. [Google Scholar] [CrossRef]
- Alvarez, B.; Martínez, -A.C.; Burgering, B.M.T.; Carrera, A.C. Forkhead Transcription Factors Contribute to Execution of the Mitotic Programme in Mammals. Nature 2001, 413, 744–747. [Google Scholar] [CrossRef]
- Kachaner, D.; Garrido, D.; Mehsen, H.; Normandin, K.; Lavoie, H.; Archambault, V. Coupling of Polo Kinase Activation to Nuclear Localization by a Bifunctional NLS Is Required during Mitotic Entry. Nat. Commun. 2017, 8, 1701. [Google Scholar] [CrossRef]
- Tavernier, N.; Thomas, Y.; Vigneron, S.; Maisonneuve, P.; Orlicky, S.; Mader, P.; Regmi, S.G.; van Hove, L.; Levinson, N.M.; Gasmi-Seabrook, G.; et al. Bora Phosphorylation Substitutes in Trans for T-Loop Phosphorylation in Aurora A to Promote Mitotic Entry. Nat. Commun. 2021, 12, 1899. [Google Scholar] [CrossRef]
- Raab, M.; Matthess, Y.; Raab, C.A.; Gutfreund, N.; Dötsch, V.; Becker, S.; Sanhaji, M.; Strebhardt, K. A Dimerization-Dependent Mechanism Regulates Enzymatic Activation and Nuclear Entry of PLK1. Oncogene 2022, 41, 372–386. [Google Scholar] [CrossRef]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Lindon, C.; Pines, J. Ordered Proteolysis in Anaphase Inactivates Plk1 to Contribute to Proper Mitotic Exit in Human Cells. J. Cell Biol. 2004, 164, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Giráldez, S.; Herrero-Ruiz, J.; Mora-Santos, M.; Japón, M.A.; Tortolero, M.; Romero, F. SCFFBXW7α Modulates the Intra-S-Phase DNA-Damage Checkpoint by Regulating Polo like Kinase-1 Stability. Oncotarget 2014, 5, 4383. [Google Scholar] [CrossRef] [PubMed]
- Giráldez, S.; Galindo-Moreno, M.; Limón-Mortés, M.C.; Cristina Rivas, A.; Herrero-Ruiz, J.; Mora-Santos, M.; Sáez, C.; Japón, M.A.; Tortolero, M.; Romero, F. G1/S Phase Progression Is Regulated by PLK1 Degradation through the CDK1/ΒTrCP Axis. FASEB J. 2017, 31, 2925–2936. [Google Scholar] [CrossRef]
- Laissue, P. The Forkhead-Box Family of Transcription Factors: Key Molecular Players in Colorectal Cancer Pathogenesis. Mol Cancer 2019, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Hannenhalli, S.; Kaestner, K.H. The Evolution of Fox Genes and Their Role in Development and Disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.F.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead Box Proteins: Tuning Forks for Transcriptional Harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox Transcription Factors: From Development to Disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef]
- Kaestner, K.H.; Knöchel, W.; Martínez, D.E. Unified Nomenclature for the Winged Helix/Forkhead Transcription Factors. Genes Dev. 2000, 14, 142–146. [Google Scholar] [CrossRef]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-Crystal Structure of the HNF-3/Fork Head DNA-Recognition Motif Resembles Histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Georges, A.B.; Benayoun, B.A.; Caburet, S.; Veitia, R.A. Generic Binding Sites, Generic DNA-binding Domains: Where Does Specific Promoter Recognition Come From? J. Fed. Am. Soc. Exp. Biol. 2010, 24, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO Code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the Master Regulator FOXM1 in Cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Wang, J.; Malovannaya, A.; Xi, Y.; Li, W.; Guerra, R.; Hawke, D.H.; Qin, J.; Chen, J. Proteomic Analyses Reveal Distinct Chromatin-associated and Soluble Transcription Factor Complexes. Mol. Syst. Biol. 2015, 11, 775. [Google Scholar] [CrossRef]
- Westendorf, J.M.; Rao, P.N.; Gerace, L. Cloning of CDNAs for M-Phase Phosphoproteins Recognized by the MPM2 Monoclonal Antibody and Determination of the Phosphorylated Epitope. Proc. Natl. Acad. Sci. USA 1994, 91, 714–718. [Google Scholar] [CrossRef]
- Suzuki, K.; Sako, K.; Akiyama, K.; Isoda, M.; Senoo, C.; Nakajo, N.; Sagata, N. Identification of Non-Ser/Thr-Pro Consensus Motifs for Cdk1 and Their Roles in Mitotic Regulation of C2H2 Zinc Finger Proteins and Ect2. Sci. Rep. 2015, 5, 7929. [Google Scholar] [CrossRef]
- Ye, H.; Kelly, T.F.; Samadani, U.; Lim, L.; Rubio, S.; Overdier, D.G.; Roebuck, K.A.; Costa, R.H. Hepatocyte Nuclear Factor 3/Fork Head Homolog 11 Is Expressed in Proliferating Epithelial and Mesenchymal Cells of Embryonic and Adult Tissues. Mol. Cell Biol. 1997, 17, 1626–1641. [Google Scholar] [CrossRef]
- Korver, W.; Roose, J.; Clevers, H. The Winged-Helix Transcription Factor Trident Is Expressed in Cycling Cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef]
- Yao, K.M.; Sha, M.; Lu, Z.; Wong, G.G. Molecular Analysis of a Novel Winged Helix Protein, WIN. Expression Pattern, DNA Binding Property, and Alternative Splicing within the DNA Binding Domain. J. Biol. Chem. 1997, 272, 19827–19836. [Google Scholar] [CrossRef]
- Barger, C.J. FOXM1 Expression and Contribution to Genomic Instability and Chemoresistance in High-Grade Serous Ovarian Cancer. Ph.D. Thesis, University of Nebraska, Lincoln, NE, USA, 2018. [Google Scholar]
- Kalin, T.V.; Ustiyan, V.; Kalinichenko, V.V. Multiple Faces of FoxM1 Transcription Factor: Lessons from Transgenic Mouse Models. Cell Cycle 2011, 10, 396–405. [Google Scholar] [CrossRef]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Krupczak-Hollis, K.; Wang, X.; Kalinichenko, V.V.; Gusarova, G.A.; Wang, I.C.; Dennewitz, M.B.; Yoder, H.M.; Kiyokawa, H.; Kaestner, K.H.; Costa, R.H. The Mouse Forkhead Box M1 Transcription Factor Is Essential for Hepatoblast Mitosis and Development of Intrahepatic Bile Ducts and Vessels during Liver Morphogenesis. Dev. Biol. 2004, 276, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Schilham, M.W.; Moerer, P.; van den Hoff, M.J.; Dam, K.; Lamers, W.H.; Medema, R.H.; Clevers, H. Uncoupling of S Phase and Mitosis in Cardiomyocytes and Hepatocytes Lacking the Winged-Helix Transcription Factor Trident. Curr. Biol. 1998, 8, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Kim, I.M.; Petrovic, V.; Malin, D.; Wang, I.C.; Kalin, T.V.; Meliton, L.; Zhao, Y.Y.; Ackerson, T.; Qin, Y.; et al. Myocardium Defects and Ventricular Hypoplasia in Mice Homozygous Null for the Forkhead Box M1 Transcription Factor. Dev. Dyn. 2007, 236, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-C.; Chen, Y.-J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol. Cell Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef]
- Wang, I.-C.; Meliton, L.; Tretiakova, M.; Costa, R.H.; Kalinichenko, V.V.; Kalin, T.V. Transgenic Expression of the Forkhead Box M1 Transcription Factor Induces Formation of Lung Tumors. Oncogene 2008, 27, 4137–4149. [Google Scholar] [CrossRef]
- Tian, J.-H.; Mu, L.-J.; Wang, M.-Y.; Zeng, J.; Long, Q.-Z.; Guan, B.; Wang, W.; Jiang, Y.M.; Bai, X.-J.; Du, Y.-F. FOXM1-Dependent Transcriptional Regulation of EZH2 Induces Proliferation and Progression in Prostate Cancer. Anticancer Agents Med. Chem. 2020, 20, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, B.; Wang, Y.U.; Zhang, M.; Fu, S.; Gao, H.; Peng, R.; Zhang, L.; Tang, J. Increased FoxM1 Expression Is a Target for Metformin in the Suppression of EMT in Prostate Cancer. Int. J. Mol. Med. 2014, 33, 1514–1522. [Google Scholar] [CrossRef]
- Park, H.J.; Gusarova, G.; Wang, Z.; Carr, J.R.; Li, J.; Kim, K.H.; Qiu, J.; Park, Y.D.; Williamson, P.R.; Hay, N.; et al. Deregulation of FoxM1b Leads to Tumour Metastasis. EMBO Mol. Med. 2011, 3, 34. [Google Scholar] [CrossRef]
- Peake, B.F.; Eze, S.M.; Yang, L.; Castellino, R.C.; Nahta, R. Growth Differentiation Factor 15 Mediates Epithelial Mesenchymal Transition and Invasion of Breast Cancers through IGF-1R-FoxM1 Signaling. Oncotarget 2017, 8, 94406. [Google Scholar] [CrossRef]
- Liu, M.; Dai, B.; Kang, S.H.; Ban, K.; Huang, F.J.; Lang, F.F.; Aldape, K.D.; Xie, T.X.; Pelloski, C.E.; Xie, K.; et al. FoxM1B Is Overexpressed in Human Glioblastomas and Critically Regulates the Tumorigenicity of Glioma Cells. Cancer Res. 2006, 66, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Kang, S.H.; Gong, W.; Liu, M.; Aldape, K.D.; Sawaya, R.; Huang, S. Aberrant FoxM1B Expression Increases Matrix Metalloproteinase-2 Transcription and Enhances the Invasion of Glioma Cells. Oncogene 2007, 26, 6212–6219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, L.; Du, Y.; Zheng, H.; Zhang, P.; Sun, Y.; Wang, Y.; Chen, J.; Ding, P.; Wang, N.; et al. A Novel FOXM1 Isoform, FOXM1D, Promotes Epithelial-Mesenchymal Transition and Metastasis through ROCKs Activation in Colorectal Cancer. Oncogene 2017, 36, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, N.; Dai, B.; Liu, M.; Sawaya, R.; Xie, K.; Huang, S. FoxM1B Transcriptionally Regulates Vascular Endothelial Growth Factor Expression and Promotes the Angiogenesis and Growth of Glioma Cells. Cancer Res. 2008, 68, 8742. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Balli, D.; Ustiyan, V.; Fulford, L.; Hiller, A.; Misetic, V.; Zhang, Y.; Paluch, A.M.; Waltz, S.E.; Kasper, S.; et al. Foxm1 Expression in Prostate Epithelial Cells Is Essential for Prostate Carcinogenesis. J. Biol. Chem. 2013, 288, 22541. [Google Scholar] [CrossRef] [PubMed]
- Karadedou, C.T.; Gomes, A.R.; Chen, J.; Petkovic, M.; Ho, K.K.; Zwolinska, A.K.; Feltes, A.; Wong, S.Y.; Chan, K.Y.K.; Cheung, Y.N.; et al. FOXO3a Represses VEGF Expression through FOXM1-Dependent and -Independent Mechanisms in Breast Cancer. Oncogene 2012, 31, 1845–1858. [Google Scholar] [CrossRef]
- Yan, Q.; Fang, X.; Li, C.; Lan, P.; Guan, X. Oncofetal Proteins and Cancer Stem Cells. Essays Biochem. 2022, 66, 423–433. [Google Scholar] [CrossRef]
- Fu, Z.; Cao, X.; Yang, Y.; Song, Z.; Zhang, J.; Wang, Z. Upregulation of FoxM1 by MnSOD Overexpression Contributes to Cancer Stem-like Cell Characteristics in the Lung Cancer H460 Cell Line. Technol. Cancer Res. Treat. 2018, 17, 1533033818789635. [Google Scholar] [CrossRef]
- Sher, G.; Masoodi, T.; Patil, K.; Akhtar, S.; Kuttikrishnan, S.; Ahmad, A.; Uddin, S. Dysregulated FOXM1 Signaling in the Regulation of Cancer Stem Cells. Semin. Cancer Biol. 2022, 86, 107–121. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W.F. FOXM1: An Emerging Master Regulator of DNA Damage Response and Genotoxic Agent Resistance. Biochim. Biophys. Acta 2014, 1839, 1316–1322. [Google Scholar] [CrossRef]
- Wierstra, I.; Alves, J. Transcription Factor FOXM1c Is Repressed by RB and Activated by Cyclin D1/Cdk4. Biol. Chem. 2006, 387, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Brison, C.M.; Nerli, S.; Arsenault, H.E.; McShan, A.C.; Chen, E.; Lee, H.W.; Benanti, J.A.; Sgourakis, N.G.; Rubin, S.M. An Order-to-Disorder Structural Switch Activates the Foxm1 Transcription Factor. Elife 2019, 8, e46131. [Google Scholar] [CrossRef] [PubMed]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead Box M1B Transcriptional Activity Requires Binding of Cdk-Cyclin Complexes for Phosphorylation-Dependent Recruitment of P300/CBP Coactivators. Mol. Cell Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef] [PubMed]
- Lüscher-Firzlaff, J.M.; Lilischkis, R.; Lüscher, B. Regulation of the Transcription Factor FOXM1c by Cyclin E/CDK2. FEBS Lett. 2006, 580, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.T.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.R.; Medema, R.H. Activation of FoxM1 during G2 Requires Cyclin A/Cdk-Dependent Relief of Autorepression by the FoxM1 N-Terminal Domain. Mol. Cell Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, C.; Wu, J.; Elsayed, Z.; Fu, Z. Polo-like Kinase 1-Mediated Phosphorylation of Forkhead Box Protein M1b Antagonizes Its SUMOylation and Facilitates Its Mitotic Function. J. Biol. Chem. 2015, 290, 3708–3719. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D.J. FOXOs, Cancer and Regulation of Apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef]
- Anderson, M.J.; Viars, C.S.; Czekay, S.; Cavenee, W.K.; Arden, K.C. Cloning and Characterization of Three Human Forkhead Genes That Comprise an FKHR-like Gene Subfamily. Genomics 1998, 47, 187–199. [Google Scholar] [CrossRef]
- Furuyama, T.; Nakazawa, T.; Nakano, I.; Mori, N. Identification of the Differential Distribution Patterns of MRNAs and Consensus Binding Sequences for Mouse DAF-16 Homologues. Biochem. J. 2000, 349, 629–634. [Google Scholar] [CrossRef]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs Are Lineage-Restricted Redundant Tumor Suppressors and Critical Regulators of Endothelial Cell Homeostasis. Cell 2007, 128, 323. [Google Scholar] [CrossRef]
- Obsil, T.; Obsilova, V. Structure/Function Relationships Underlying Regulation of FOXO Transcription Factors. Oncogene 2008, 27, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- van der Heide, L.P.; Hoekman, M.F.M.; Smidt, M.P. The Ins and Outs of FoxO Shuttling: Mechanisms of FoxO Translocation and Transcriptional Regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Blee, A.M.; Wang, D.; An, J.; Pan, Y.; Yan, Y.; Ma, T.; He, Y.; Dugdale, J.; Hou, X.; et al. Loss of FOXO1 Cooperates with TMPRSS2–ERG Overexpression to Promote Prostate Tumorigenesis and Cell Invasion. Cancer Res. 2017, 77, 6524–6537. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Lee, S.; Maeda, M.; Kumagai-Takei, N.; Nishimura, Y.; Otsuki, T. FoxO1 Regulates Apoptosis Induced by Asbestos in the MT-2 Human T-Cell Line. J. Immunotoxicol. 2016, 13, 620–627. [Google Scholar] [CrossRef]
- Modur, V.; Nagarajan, R.; Evers, B.M.; Milbrandt, J. FOXO Proteins Regulate Tumor Necrosis Factor-Related Apoptosis Inducing Ligand Expression: Implications for PTEN Mutation in Prostate Cancer. J. Biol. Chem. 2002, 277, 47928–47937. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Nakamura, N.; Sansal, I.; Bergeron, L.; Sellers, W.R. A Novel Mechanism of Gene Regulation and Tumor Suppression by the Transcription Factor FKHR. Cancer Cell 2002, 2, 81–91. [Google Scholar] [CrossRef]
- Nakamura, N.; Ramaswamy, S.; Vazquez, F.; Signoretti, S.; Loda, M.; Sellers, W.R. Forkhead Transcription Factors Are Critical Effectors of Cell Death and Cell Cycle Arrest Downstream of PTEN. Mol. Cell Biol. 2000, 20, 8969–8982. [Google Scholar] [CrossRef]
- Ju, Y.; Xu, T.; Zhang, H.; Yu, A. FOXO1-Dependent DNA Damage Repair Is Regulated by JNK in Lung Cancer Cells. Int. J. Oncol. 2014, 44, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Mokry, M.; de Wit, E.; Smits, L.M.; Polderman, P.E.; van Triest, M.H.; van Boxtel, R.; Schulze, A.; de Laat, W.; Cuppen, E.; et al. Genome-Wide Analysis of FOXO3 Mediated Transcription Regulation through RNA Polymerase II Profiling. Mol. Syst. Biol. 2013, 9, 638. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox Regulation of FoxO Transcription Factors. Redox. Biol. 2015, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yalcin, S.; Lee, D.F.; Yeh, T.Y.J.; Lee, S.M.; Su, J.; Mungamuri, S.K.; Rimmelé, P.; Kennedy, M.; Sellers, R.; et al. FOXO1 Is an Essential Regulator of Pluripotency in Human Embryonic Stem Cells. Nat. Cell Biol. 2011, 13, 1099. [Google Scholar] [CrossRef]
- Tikhanovich, I.; Cox, J.; Weinman, S.A. FOXO Transcription Factors in Liver Function and Disease. J. Gastroenterol. Hepatol. 2013, 28, 131. [Google Scholar] [CrossRef]
- Paik, J.H.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.S.; Zheng, H.; Ying, H.; Mahoney, J.; et al. FoxOs Cooperatively Regulate Diverse Pathways Governing Neural Stem Cell Homeostasis. Cell Stem Cell 2009, 5, 553. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β–FOXO Signalling Maintains Leukaemia-Initiating Cells in Chronic Myeloid Leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef]
- Yu, J.M.; Sun, W.; Wang, Z.H.; Liang, X.; Hua, F.; Li, K.; Lv, X.X.; Zhang, X.W.; Liu, Y.Y.; Yu, J.J.; et al. TRIB3 Supports Breast Cancer Stemness by Suppressing FOXO1 Degradation and Enhancing SOX2 Transcription. Nat. Commun. 2019, 10, 5720. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chauhan, A.S.; Zhuang, L.; Gan, B. FoxO Transcription Factors in Cancer Metabolism. Semin Cancer Biol. 2018, 50, 65–76. [Google Scholar] [CrossRef]
- Dong, T.; Zhang, Y.; Chen, Y.; Liu, P.; An, T.; Zhang, J.; Yang, H.; Zhu, W.; Yang, X. FOXO1 Inhibits the Invasion and Metastasis of Hepatocellular Carcinoma by Reversing ZEB2-Induced Epithelial-Mesenchymal Transition. Oncotarget 2017, 8, 1713. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, R.; Ye, N.; Liu, C.; Li, X.; Guo, X.; Zhang, Z.; Li, X.; Yao, Y.; Jiang, X. FOXO1 Inhibits Tumor Cell Migration via Regulating Cell Surface Morphology in Non-Small Cell Lung Cancer Cells. Cell. Physiol. Biochem. 2018, 48, 138–148. [Google Scholar] [CrossRef]
- Zhang, X.; Zhuang, T.; Liang, Z.; Li, L.; Xue, M.; Liu, J.; Liang, H. Breast Cancer Suppression by Aplysin Is Associated with Inhibition of PI3K/AKT/FOXO3a Pathway. Oncotarget 2017, 8, 63934. [Google Scholar] [CrossRef]
- Fang, L.; Wang, H.; Zhou, L.; Yu, D. Akt-FOXO3a Signaling Axis Dysregulation in Human Oral Squamous Cell Carcinoma and Potent Efficacy of FOXO3a-Targeted Gene Therapy. Oral Oncol. 2011, 47, 16–21. [Google Scholar] [CrossRef]
- Yang, J.Y.; Xia, W.; Hu, M.C.T. Ionizing Radiation Activates Expression of FOXO3a, Fas Ligand, Bim, and Induces Cell Apoptosis. Int. J. Oncol. 2006, 29, 648. [Google Scholar] [CrossRef]
- Yamamura, Y.; Wei, L.L.; Inoue, K.I.; Ida, H.; Ito, Y. RUNX3 Cooperates with FoxO3a to Induce Apoptosis in Gastric Cancer Cells. J. Biol. Chem. 2006, 281, 5267–5276. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Chen, Y.; Zhang, Q.; Guo, Y.; Huang, Z.; Dai, L.; Cao, S. 8-Bromo-7-Methoxychrysin Induces Apoptosis by Regulating Akt/FOXO3a Pathway in Cisplatin-Sensitive and Resistant Ovarian Cancer Cells. Mol. Med. Rep. 2015, 12, 5108. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Song, Y.H.; Yokomizo, A.; Kiyoshima, K.; Tada, Y.; Uchino, H.; Uchiumi, T.; Inokuchi, J.; Oda, Y.; Kuroiwa, K.; et al. Foxo3a Suppression of Urothelial Cancer Invasiveness through Twist1, Y-Box–Binding Protein 1, and E-Cadherin Regulation. Clin. Cancer Res. 2010, 16, 5654–5663. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Bhaskaran, N.; Maclennan, G.T.; Gupta, S. Deregulation of FoxO3a Accelerates Prostate Cancer Progression in TRAMP Mice. Prostate 2013, 73, 1517. [Google Scholar] [CrossRef]
- Hornsveld, M.; Dansen, T.B.; Derksen, P.W.; Burgering, B.M.T. Re-Evaluating the Role of FOXOs in Cancer. Semin. Cancer Biol. 2018, 50, 90–100. [Google Scholar] [CrossRef]
- Chen, Y.H.; Li, C.L.; Chen, W.J.; Liu, J.; Wu, H.T. Diverse Roles of FOXO Family Members in Gastric Cancer. World J. Gastrointest. Oncol. 2021, 13, 1382. [Google Scholar] [CrossRef] [PubMed]
- van der Vos, K.E.; Coffer, P.J. FOXO-Binding Partners: It Takes Two to Tango. Oncogene 2008, 27, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Burgering, B.M.T. FOXOs: Masters of the Equilibrium. FEBS J. 2021, 289, 7918–7939. [Google Scholar] [CrossRef] [PubMed]
- Mattila, J.; Kallijärvi, J.; Puig, O. RNAi Screening for Kinases and Phosphatases Identifies FoxO Regulators. Proc. Natl. Acad. Sci. USA 2008, 105, 14873–14878. [Google Scholar] [CrossRef]
- Bucur, O.; Stancu, A.L.; Muraru, M.S.; Melet, A.; Petrescu, S.M.; Khosravi-Far, R. PLK1 Is a Binding Partner and a Negative Regulator of FOXO3 Tumor Suppressor. Discoveries 2014, 2, e16. [Google Scholar] [CrossRef]
- Geng, F.-S.; de la Calle-Mustienes, E.; Gómez-Skarmeta, J.L.; Lister, R.; Bogdanovic, O. Depletion of Foxk Transcription Factors Causes Genome-Wide Transcriptional Misregulation and Developmental Arrest in Zebrafish Embryos. MicroPubl. Biol. 2020, 2020. [Google Scholar] [CrossRef]
- Garry, D.J.; Maeng, G.; Garry, M.G. Foxk1 Regulates Cancer Progression. Ann. Transl. Med. 2020, 8, 1041. [Google Scholar] [CrossRef]
- Bassel-Duby, R.; Hernandez, M.D.; Yang, Q.; Rochelle, J.M.; Seldin, M.F.; Williams, R.S. Myocyte Nuclear Factor, a Novel Winged-Helix Transcription Factor under Both Developmental and Neural Regulation in Striated Myocytes. Mol. Cell Biol. 1994, 14, 4596–4605. [Google Scholar] [CrossRef]
- Garry, D.J.; Yang, Q.; Bassel-Duby, R.; Williams, R.S. Persistent Expression of MNF Identifies Myogenic Stem Cells in Postnatal Muscles. Dev. Biol. 1997, 188, 280–294. [Google Scholar] [CrossRef]
- Huang, J.T.J.; Lee, V. Identification and Characterization of a Novel Human FOXK1 Gene in Silico. Int. J. Oncol. 2004, 25, 751–757. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-Based Map of the Human Proteome. Science 2015, 347, 6220. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, W.; Ge, H.; Ponnusamy, M.; Wang, Q.; Hao, X.; Wu, W.; Zhang, Y.; Yu, W.; Ao, X.; et al. FOXK Transcription Factors: Regulation and Critical Role in Cancer. Cancer Lett. 2019, 458, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Garry, D.J. Sin3 Interacts with Foxk1 and Regulates Myogenic Progenitors. Mol. Cell Biochem. 2012, 366, 251–258. [Google Scholar] [CrossRef]
- Garry, D.J.; Meeson, A.; Elterman, J.; Zhao, Y.; Yang, P.; Bassel-Duby, R.; Williams, R.S. Myogenic Stem Cell Function Is Impaired in Mice Lacking the Forkhead/Winged Helix Protein MNF. Proc. Natl. Acad. Sci. USA 2000, 97, 5421. [Google Scholar] [CrossRef]
- Ji, Z.G.; Jiang, H.T.; Zhang, P.S. FOXK1 Promotes Cell Growth through Activating Wnt/β-Catenin Pathway and Emerges as a Novel Target of MiR-137 in Glioma. Am. J. Transl. Res. 2018, 10, 1784–1792. [Google Scholar]
- Peng, Y.; Zhang, P.; Huang, X.; Yan, Q.; Wu, M.; Xie, R.; Wu, Y.; Zhang, M.; Nan, Q.; Zhao, J.; et al. Direct Regulation of FOXK1 by C-Jun Promotes Proliferation, Invasion and Metastasis in Gastric Cancer Cells. Cell Death Dis. 2016, 7, e2480. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, J.; Tang, W.; Zhan, X.; Li, Y.; Peng, Y.; Huang, X.; Bai, Y.; Zhao, J.; Li, A.; et al. FOXK1 Interaction with FHL2 Promotes Proliferation, Invasion and Metastasis in Colorectal Cancer. Oncogenesis 2016, 5, e271. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. MTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960.e4. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Cai, W.; Wang, C.H.; Cederquist, C.T.; Damasio, M.; Homan, E.P.; Batista, T.; Ramirez, A.K.; Gupta, M.K.; Steger, M.; et al. FoxK1 and FoxK2 in Insulin Regulation of Cellular and Mitochondrial Metabolism. Nat. Commun. 2019, 10, 1582. [Google Scholar] [CrossRef]
- Bowman, C.J.; Ayer, D.E.; Dynlacht, B.D. Foxk Proteins Repress the Initiation of Starvation-Induced Atrophy and Autophagy Programs. Nat. Cell Biol. 2014, 16, 1202–1214. [Google Scholar] [CrossRef]
- Nakatsumi, H.; Matsumoto, M.; Nakayama, K.I. Noncanonical Pathway for Regulation of CCL2 Expression by an MTORC1-FOXK1 Axis Promotes Recruitment of Tumor-Associated Macrophages. Cell Rep. 2017, 21, 2471–2486. [Google Scholar] [CrossRef]
- Nakatsumi, H.; Oka, T.; Higa, T.; Shirane, M.; Nakayama, K.I. Nuclear–Cytoplasmic Shuttling Protein PP2AB56 Contributes to MTORC1-Dependent Dephosphorylation of FOXK1. Genes Cells 2018, 23, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, P.; Lee, C.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Sharrocks, A.D.; Haines, D.S.; Reddy, E.P. JNK-Associated Leucine Zipper Protein Functions as a Docking Platform for Polo-like Kinase 1 and Regulation of the Associating Transcription Factor Forkhead Box Protein K1. J. Biol. Chem. 2015, 290, 29617–29628. [Google Scholar] [CrossRef] [PubMed]
- Pietila, M.; Vijay, G.V.; Soundararajan, R.; Yu, X.; Symmans, W.F.; Sphyris, N.; Mani, S.A. FOXC2 Regulates the G2/M Transition of Stem Cell-Rich Breast Cancer Cells and Sensitizes Them to PLK1 Inhibition. Sci. Rep. 2016, 6, 23070. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Miyazaki, T.; Fujisawa, K.; Nasu, K.; Hamanaka, R.; Miyakawa, I. Expression of Polo-like Kinase in Ovarian Cancer Is Associated with Histological Grade and Clinical Stage. Cancer Lett. 2001, 164, 41–49. [Google Scholar] [CrossRef]
- Weichert, W.; Schmidt, M.; Gekeler, V.; Denkert, C.; Stephan, C.; Jung, K.; Loening, S.; Dietel, M.; Kristiansen, G. Polo-like Kinase 1 Is Overexpressed in Prostate Cancer and Linked to Higher Tumor Grades. Prostate 2004, 60, 240–245. [Google Scholar] [CrossRef]
- Weichert, W.; Ullrich, A.; Schmidt, M.; Gekeler, V.; Noske, A.; Niesporek, S.; Buckendahl, A.-C.; Dietel, M.; Denkert, C. Expression Patterns of Polo-like Kinase 1 in Human Gastric Cancer. Cancer Sci. 2006, 97, 271–276. [Google Scholar] [CrossRef]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.-M.; Rübsamen-Waigmann, H.; Strebhardt, K. Prognostic Significance of Polo-like Kinase (PLK) Expression in Non-Small Cell Lung Cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef]
- Knecht, R.; Elez, R.; Oechler, M.; Solbach, C.; Ilberg, C.V.; Strebhardt, K. Prognostic Significance of Polo-like Kinase (PLK) Expression in Squamous Cell Carcinomas of the Head and Neck. Cancer Res. 1999, 59, 2794–2797. [Google Scholar]
- Knecht, R.; Oberhauser, C.; Strebhardt, K. PLK (Polo-like Kinase), a New Prognostic Marker for Oropharyngeal Carcinomas. Int. J. Cancer 2000, 89, 535–536. [Google Scholar] [CrossRef]
- Kong, Y.; Allison, D.B.; Zhang, Q.; He, D.; Li, Y.; Mao, F.; Li, C.; Li, Z.; Zhang, Y.; Wang, J.; et al. The Kinase PLK1 Promotes the Development of Kras/Tp53-Mutant Lung Adenocarcinoma through Transcriptional Activation of the Receptor RET. Sci. Signal. 2022, 15, 754. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Marshall, C.B.; Yamamoto, K.; Li, G.-Y.; Plevin, M.J.; You, H.; Mak, T.W.; Ikura, M. Biochemical and Structural Characterization of an Intramolecular Interaction in FOXO3a and Its Binding with P53. J. Mol. Biol. 2008, 384, 590–603. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Yamamoto, K.; Mak, T.W. Regulation of Transactivation-Independent Proapoptotic Activity of P53 by FOXO3a. Proc. Natl. Acad. Sci. USA 2006, 103, 9051–9056. [Google Scholar] [CrossRef] [PubMed]
- Kurinna, S.; Stratton, S.A.; Coban, Z.; Schumacher, J.M.; Grompe, M.; Duncan, A.W.; Barton, M.C. P53 Regulates a Mitotic Transcription Program and Determines Ploidy in Normal Mouse Liver. Hepatology 2013, 57, 2004–2013. [Google Scholar] [CrossRef]
- Barsotti, A.M.; Prives, C. Pro-Proliferative FoxM1 Is a Target of P53-Mediated Repression. Oncogene 2009, 28, 4295–4305. [Google Scholar] [CrossRef]
- McKenzie, L.; King, S.; Marcar, L.; Nicol, S.; Dias, S.S.; Schumm, K.; Robertson, P.; Bourdon, J.-C.; Perkins, N.; Fuller-Pace, F.; et al. P53-Dependent Repression of Polo-like Kinase-1 (PLK1). Cell Cycle 2010, 9, 4200–4212. [Google Scholar] [CrossRef]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic Value of FOXM1 in Solid Tumors: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 32308. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Zhang, Y.; Jia, L.; Zhang, Y.; Ji, W.; Li, H. Higher Expression of FOXOs Correlates to Better Prognosis of Bladder Cancer. Oncotarget 2017, 8, 96322. [Google Scholar] [CrossRef]
- Ide, H.; Jiang, G.; Mizushima, T.; Fujita, K.; Inoue, S.; Yamaguchi, S.; Fushimi, H.; Nonomura, N.; Miyamoto, H. Forkhead Box O1 as an Indicator of Prognosis Is Inactivated in Urothelial Carcinoma of the Upper Urinary Tract. Oncol. Lett. 2019, 17, 487. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, Z.; Qin, Y.; Wang, X.; Sun, L.; Zhang, Y.; Gong, L.; Wu, S.; Han, S.; Tang, Y.; et al. FOXO1 Promotes Tumor Progression by Increased M2 Macrophage Infiltration in Esophageal Squamous Cell Carcinoma. Theranostics 2020, 10, 11548. [Google Scholar] [CrossRef]
- Sorensen, P.H.B.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR Gene Fusions Are Prognostic Indicators in Alveolar Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Shou, Z.; Lin, L.; Liang, J.; Li, J.L.; Chen, H.Y. Expression and Prognosis of FOXO3a and HIF-1α in Nasopharyngeal Carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Fondevila, F.; Fernández-Palanca, P.; Méndez-Blanco, C.; Payo-Serafín, T.; Lozano, E.; Marin, J.J.G.; González-Gallego, J.; Mauriz, J.L. Association of FOXO3 Expression with Tumor Pathogenesis, Prognosis and Clinicopathological Features in Hepatocellular Carcinoma: A Systematic Review with Meta-Analysis. Cancers 2021, 13, 5349. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gong, M.; Zhao, Y.; Zhao, X.; Li, Q. FOXK1 Facilitates Cell Proliferation through Regulating the Expression of P21, and Promotes Metastasis in Ovarian Cancer. Oncotarget 2017, 8, 70441–70451. [Google Scholar] [CrossRef]
- Cao, H.; Chu, X.; Wang, Z.; Guo, C.; Shao, S.; Xiao, J.; Zheng, J.; Zhang, D. High FOXK1 Expression Correlates with Poor Outcomes in Hepatocellular Carcinoma and Regulates Stemness of Hepatocellular Carcinoma Cells. Life Sci. 2019, 228, 128–134. [Google Scholar] [CrossRef]
- Elsayed, I.; Wang, X. PLK1 Inhibition in Cancer Therapy: Potentials and Challenges. Future Med. Chem. 2019, 11, 1383–1386. [Google Scholar] [CrossRef]
- Wu, C.-P.; Hsieh, C.-H.; Hsiao, S.-H.; Luo, S.-Y.; Su, C.-Y.; Li, Y.-Q.; Huang, Y.-H.; Huang, C.-W.; Hsu, S.-C. Human ATP-Binding Cassette Transporter ABCB1 Confers Resistance to Volasertib (BI 6727), a Selective Inhibitor of Polo-like Kinase 1. Mol. Pharm. 2015, 12, 3885–3895. [Google Scholar] [CrossRef]
- Reindl, W.; Yuan, J.; Krämer, A.; Strebhardt, K.; Berg, T. Inhibition of Polo-like Kinase 1 by Blocking Polo-Box Domain-Dependent Protein-Protein Interactions. Chem. Biol. 2008, 15, 459–466. [Google Scholar] [CrossRef]
- Archambault, V.; Normandin, K. Several Inhibitors of the Plk1 Polo-Box Domain Turn out to Be Non-Specific Protein Alkylators. Cell Cycle 2017, 16, 1220–1224. [Google Scholar] [CrossRef]
- Scharow, A.; Raab, M.; Saxena, K.; Sreeramulu, S.; Kudlinzki, D.; Gande, S.; Dötsch, C.; Kurunci-Csacsko, E.; Klaeger, S.; Kuster, B.; et al. Optimized Plk1 PBD Inhibitors Based on Poloxin Induce Mitotic Arrest and Apoptosis in Tumor Cells. ACS Chem. Biol. 2015, 10, 2570–2579. [Google Scholar] [CrossRef]
- Rubner, S.; Scharow, A.; Schuber, S.; Berg, T. Selective Degradation of Polo-like Kinase 1 by a Hydrophobically Tagged Inhibitor of the Polo-Box Domain. Angew. Chem. Int. Ed. Engl. 2018, 57, 17043–17047. [Google Scholar] [CrossRef]
- Hymel, D.; Tsuji, K.; Grant, R.A.; Chingle, R.M.; Kunciw, D.L.; Yaffe, M.B.; Burke, T.R. Design and Synthesis of a New Orthogonally Protected Glutamic Acid Analog and Its Use in the Preparation of High Affinity Polo-like Kinase 1 Polo-Box Domain—Binding Peptide Macrocycles. Org. Biomol. Chem. 2021, 19, 7854. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, A.B.; Jones, K.L.; Harling, J.D. The Therapeutic Potential of PROTACs. Expert Opin. Ther. Pat. 2021, 31, 1–24. [Google Scholar] [CrossRef]
- Martín-Acosta, P.; Xiao, X. PROTACs to Address the Challenges Facing Small Molecule Inhibitors. Eur. J. Med. Chem. 2021, 210, 112993. [Google Scholar] [CrossRef]
- Mu, X.; Bai, L.; Xu, Y.; Wang, J.; Lu, H. Protein Targeting Chimeric Molecules Specific for Dual Bromodomain 4 (BRD4) and Polo-like Kinase 1 (PLK1) Proteins in Acute Myeloid Leukemia Cells. Biochem. Biophys. Res. Commun. 2020, 521, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10, 3472. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Ikeda, Y.; Kunugi, Y.; Kurosaki, A.; Imai, Y.; Kohyama, S.; Nagao, S.; Kozawa, E.; Yoshida, K.; Tsunoda, T.; et al. Phase I Study of Multiple Epitope Peptide Vaccination in Patients With Recurrent or Persistent Cervical Cancer. J. Immunother. 2018, 41, 201–207. [Google Scholar] [CrossRef]
- Takeuchi, S.; Kagabu, M.; Shoji, T.; Nitta, Y.; Sugiyama, T.; Sato, J.; Nakamura, Y. Anti-Cancer Immunotherapy Using Cancer-Derived Multiple Epitope-Peptides Cocktail Vaccination Clinical Studies in Patients with Refractory/Persistent Disease of Uterine Cervical Cancer and Ovarian Cancer [Phase 2]. Oncoimmunology 2020, 9, 1838189. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Okada, K.; Omori, T.; Sugimura, K.; Miyata, H.; Ohue, M.; Kobayashi, S.; Takahashi, H.; Nakano, H.; Mochizuki, C.; et al. Multiple Therapeutic Peptide Vaccines for Patients with Advanced Gastric Cancer. Int. J. Oncol. 2017, 50, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Hosono, A.; Yoshikawa, T.; Kaneda, H.; Nitani, C.; Hara, J.; Kinoshita, Y.; Kohashi, K.; Manabe, A.; Fukutani, M.; et al. Efficacy of the NCCV Cocktail-1 Vaccine for Refractory Pediatric Solid Tumors: A Phase I Clinical Trial. Cancer Sci. 2019, 110, 3662. [Google Scholar] [CrossRef] [PubMed]
- Gheghiani, L.; Shang, S.; Fu, Z. Targeting the PLK1-FOXO1 Pathway as a Novel Therapeutic Approach for Treating Advanced Prostate Cancer. Sci. Rep. 2020, 10, 12327. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef]
- Thalhammer, V.; Lopez-Garcia, L.A.; Herrero-Martin, D.; Hecker, R.; Laubscher, D.; Gierisch, M.E.; Wachtel, M.; Bode, P.; Nanni, P.; Blank, B.; et al. PLK1 Phosphorylates PAX3-FOXO1, the Inhibition of Which Triggers Regression of Alveolar Rhabdomyosarcoma. Cancer Res. 2015, 75, 98–110. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid Biopsy Enters the Clinic—Implementation Issues and Future Challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a New Frontier of Cancer Liquid Biopsy. Mol. Cancer 2022, 21, 56. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwängler, B.; Üresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; auf dem Keller, U.; Dick, J.E.; Porse, B.T. Quantitative Single-Cell Proteomics as a Tool to Characterize Cellular Hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, X.T.R.; Gheghiani, L.; Fu, Z. The Role of Polo-Like Kinase 1 in Regulating the Forkhead Box Family Transcription Factors. Cells 2023, 12, 1344. https://doi.org/10.3390/cells12091344
Moore XTR, Gheghiani L, Fu Z. The Role of Polo-Like Kinase 1 in Regulating the Forkhead Box Family Transcription Factors. Cells. 2023; 12(9):1344. https://doi.org/10.3390/cells12091344
Chicago/Turabian StyleMoore, Xavier T. R., Lilia Gheghiani, and Zheng Fu. 2023. "The Role of Polo-Like Kinase 1 in Regulating the Forkhead Box Family Transcription Factors" Cells 12, no. 9: 1344. https://doi.org/10.3390/cells12091344