
Senescent Cells: A Therapeutic Target in Cardiovascular Diseases

 , , ,
, , ,
Abstract
:1. Introduction
2. Molecular Mechanisms of Cellular Senescence
3. Markers of Cellular Senescence
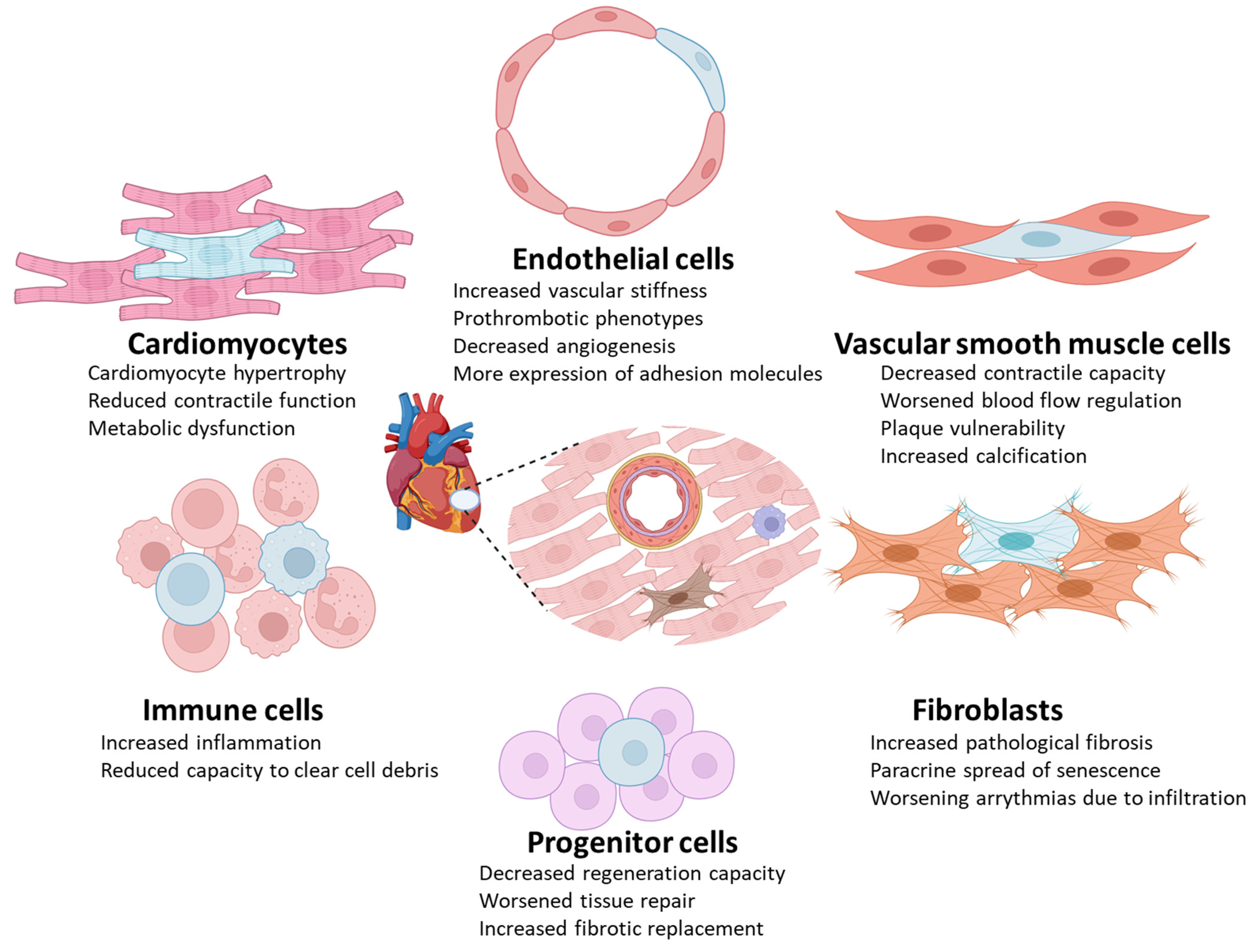
4. Detrimental Effects of Certain Types of Senescent Cells in Cardiovascular Diseases
4.1. Endothelial Cells
4.2. Vascular Smooth Muscle Cells
4.3. Cardiomyocytes
4.4. Immune Cells
4.5. Progenitor Cells
4.6. Fibroblasts
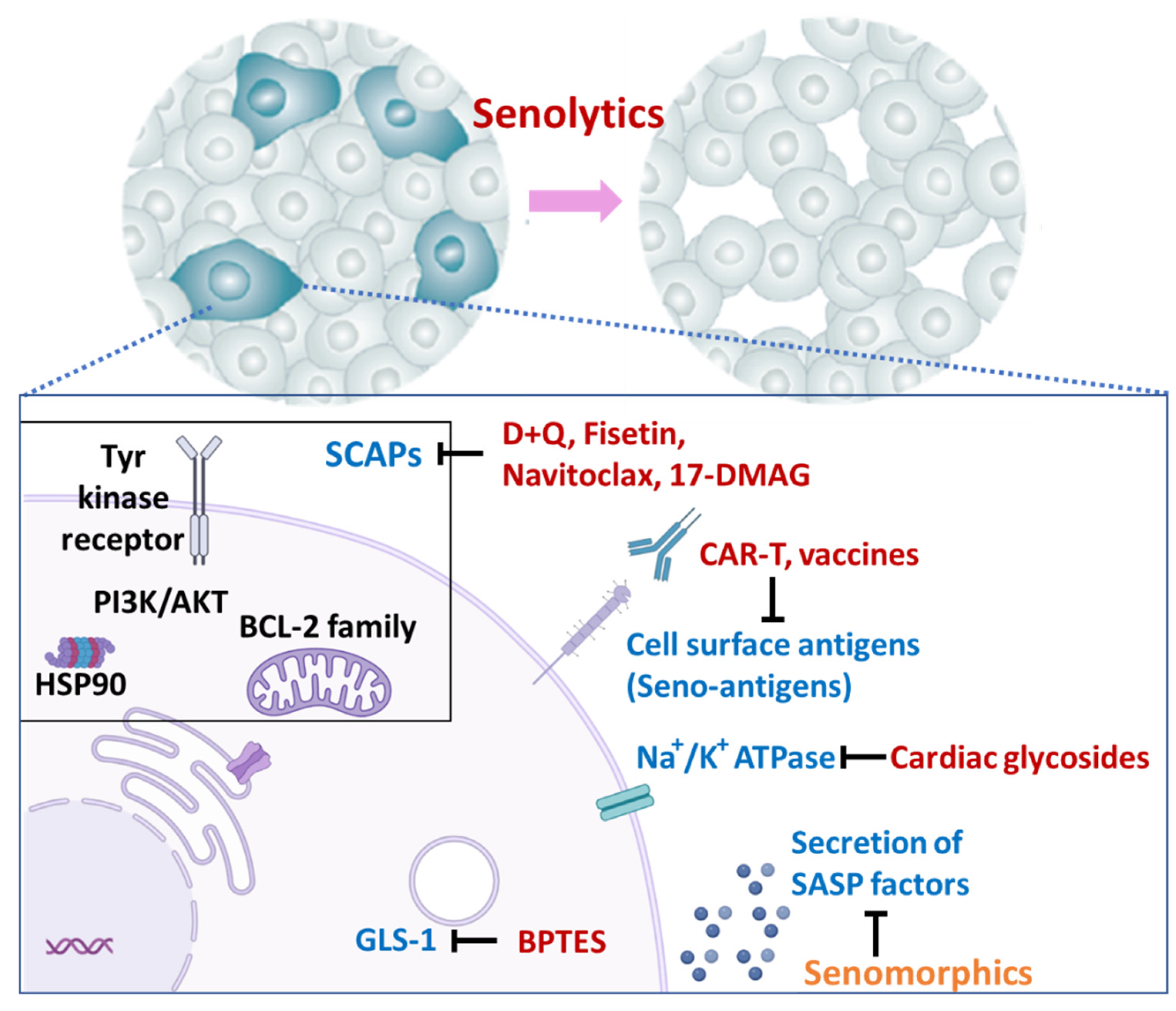
5. Therapies Targeting Senescent Cells
5.1. Elimination of Senescent Cells (Senolytics)
5.2. Other Strategies
6. Cardiovascular Diseases That Senotherapeutics May Potentially Alleviate

{kind=link}
{kind=link}
| Model Type | Senescent Cell Removal Model | Senolytic Drug | Result of Senolytic Therapy | Reference |
|---|---|---|---|---|
| Aging (mouse) | D+Q | Improved left ventricular ejection fraction | [47] | |
| INK-ATTAC | D+Q | Activated resident cardiac progenitor cells and cardiomyocyte formation | [7] | |
| INK-ATTAC | Navitoclax | Alleviated myocardial hypertrophy and fibrosis | [54] | |
| D+Q | Prolonged survival of old cardiac allografts | [261] | ||
| D+Q | Improved vascular relaxation | [47] | ||
| INK-ATTAC | D+Q | Reduced aortic calcification and improved vasomotor function | [282] | |
| High-fat diet-induced heart failure (rat) | Q | Attenuated high-fat diet-induced cardiac systolic dysfunction | [259] | |
| High-fat diet-induced heart failure (mouse) | Q | Normalized heart size and reduced cardiac fat and fibrosis | [8] | |
| High-fat diet-induced heart failure (mouse) | D+Q | Improved CPC proliferation and cardiac repair | [185] | |
| Angiotensin II (Ang II)-induced heart failure (mouse) | Navitoclax | Improved left ventricular ejection fraction | [262] | |
| Doxorubicin (DOX)-induced heart failure (mouse) | Navitoclax | Improved cardiac function | [260] | |
| Ischemia–reperfusion injury (mouse) | Navitoclax | Alleviated myocardial hypertrophy and fibrosis, improved left ventricular ejection fraction, increased myocardial vascularization | [272] | |
| Aged mice of following MI (mouse) | Navitoclax | Increased survival after myocardial infarction | [273] | |
| Ischemia and reperfusion-injury (rat) | Navitoclax | Improved cardiac function and increased angiogenesis | [270] | |
| Myocardial infarction (mouse) | D+Q | Improved cardiac function, increased regeneration | [271] | |
| AF after acute myocardial infarction (rat) | Fisetin | Attenuated atrial fibrosis and AF duration following acute myocardial infarction | [268] | |
| Isoproterenol induced AF (rat) | Q | Alleviated fibrosis and collagen deposition in atrial tissues | [267] | |
| Atherosclerosis (Ldlr KO) (mouse) | INK-ATTAC p16-3MR | Navitoclax | Reduced atherosclerotic plaque formation and instability | [162] |
| INK-ATTAC p16-3MR | Navitoclax | Improved fibrous cap thickness | [130] | |
| Atherosclerosis (ApoE KO) (mouse) | p16-3MR | Navitoclax | Reduced atherosclerotic plaque size and inflammatory cell count | [279] |
| GPNMB vaccine | Reduced atherosclerotic plaque formation and inflammation | [223] | ||
| Digoxin | Reduced atherosclerotic lesion formation and plasma lipid levels | [280] | ||
| 17-DMAG | Reduced atherosclerotic lesions and induced a more stable plaque phenotype | [281] | ||
| BPTES | Reduce atherosclerotic plaque formation | [220] | ||
| Dialysis arteriovenous fistula (mouse) | D+Q | Decreased senescent cells in dialysis arteriovenous fistula mice | [284] | |
| MCT and shunt-induced PAH (rat) | Navitoclax | Improved hemodynamic and structural changes associated with severe PAH | [108] | |
| Oxygen-induced retinopathy (mouse), eye tissue (human) | INK-ATTAC | UBX1967 | Ameliorated ischemic retinopathy | [286] |
| Angiotensin II (Ang II) administrated, aged (mouse) | D+Q | Reduced AAA size | [285] | |
| Cholesterol diet (rabbit) | Navitoclax | Reduced stenosis area of senolytic-coated stent | [25] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 53BP1 | Tumor suppressor p53-binding protein 1 |
| ACE | Angiotensin-converting enzyme |
| ARBs | Angiotensin receptor blockers |
| AFAR | American Federation for Aging Research |
| AKT1 | Threonine protein kinase |
| ALP | Alkaline phosphatase |
| AMPK | AMP-activated protein kinase |
| Ang II | Angiotensin II |
| ANGPTL2 | Angiopoietin-like protein 2 |
| ApoE | Apolipoprotein E |
| BCL-xL | B-cell lymphoma-extra large |
| BMP-2 | Bone morphogenetic protein 2 |
| BPTES | Bis-2-(5-phenyl- acetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide |
| CABG | Coronary artery bypass graft |
| CAR-T | Chimeric antigen receptor T cell |
| CCL3 | Chemokine (C-C motif) ligand 3 |
| CCN1 | CCN family member 1 |
| CCR2 | C-C motif chemokine receptor 2 |
| CF | Cardiac fibroblasts |
| CMV | Cytomegalovirus |
| COPD | Chronic obstructive pulmonary disease |
| CPC | Cardiac progenitor cell |
| CPT1 | Carnitine palmitoyltransferase I |
| CVD | Cardiovascular disease |
| D | Dasatinib |
| DAMP | Damage-associated molecular pattern |
| DNA | Deoxyribonucleic acid |
| DNA-SCARS | DNA segments with chromatin alterations reinforcing senescence |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| Edn3 | Endothelin 3 |
| EFNB1 | Ephrin B1 |
| eNOS | Endothelial nitric oxide synthase |
| EPCs | Endothelial progenitor cells |
| ET-1 | Endothelin 1 |
| FAS | TNF receptor superfamily member 6 |
| FGF | Fibroblast growth factor |
| FOXO | Forkhead box protein O |
| GDF15 | Growth differentiation factor 15 |
| GH | Growth hormone |
| GHRH | Growth hormone–releasing hormone |
| GLS1 | Glutaminase 1 |
| GPNMB | Glycoprotein nonmetastatic melanoma protein B |
| HF | Heart failure |
| HFpEF | Heart failure with preserved ejection fraction |
| HGPS | Hutchinson–Gilford progeria syndrome |
| HIF | Hypoxia-inducible factor |
| HMGB-1 | High mobility group box 1 |
| hs-CRP | High sensitivity C-reactive protein |
| HSP | Heat shock protein |
| HSP90 | Heat shock protein 90 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| ICD | International Classification of Diseases |
| IFN- γ | Interferon-gamma |
| IGF-1 | Insulin-like growth factor 1 |
| IGFBP | Insulin-like Growth Factor-binding Protein |
| IL | Interleukin |
| KGA | Kidney-type glutaminase |
| Ki-67 | Marker of proliferation Ki-67 |
| KO | Knockout |
| LDL | Low-density lipoprotein |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MI | Myocardial infarction |
| miRNAs | MicroRNAs |
| MMP | Matrix Metallopeptidase |
| mTOR | Mammalian target of rapamycin |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide dinucleotide phosphate |
| NAFLD | Non-alcoholic fatty liver disease |
| NK | Natural killer |
| NO | Nitric oxide |
| NOXs | Nicotinamide dinucleotide phosphate oxidases |
| OIS | Oncogene induced senescence |
| OPG | Osteoprotegerin |
| OPN | Osteopontin |
| OSKM | The “Yamanaka factors”, Oct4, Sox2, Klf4, and c-Myc |
| p16Ink4a | Cyclin-dependent kinase inhibitor 2A |
| p21CIP1/WAF1 | Cyclin-dependent kinase inhibitor 1 (Cdkn1a) |
| p38MAPK | p38 mitogen-activated protein kinase |
| p53 | Cellular tumor antigen p53 |
| PAH | Pulmonary arterial hypertension |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphoinositide 3-kinases |
| PPARα | Peroxisome proliferator-activated receptor α |
| Q | Quercetin |
| RAAS | Renin-angiotensin-aldosterone system |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| Runx-2 | runt-associated transcription factor 2 |
| SADs | Senescence-associated distention of satellites |
| SAHF | Senescence-associated heterochromatin foci |
| SASP | Senescence-associated secretory phenotype |
| SA-βgal | Senescence-associated beta-galactosidase |
| SCAPs | Senescent cell anti-apoptotic pathways |
| scRNA-seq | Single cell RNA-sequencing |
| SGLT2 | Sodium-Glucose Transport Protein 2 |
| SIRT | Sirtuin |
| SM-MHC | Smooth muscle myosin heavy chain |
| SOD | Superoxide dismutase |
| TAFs | Telomere-associated foci |
| TAME | Targeting Aging with Metformin |
| TEM | Transmission electron microscopy |
| TGFβ | Tumor growth factor-beta |
| TIS | Therapy-induced senescent cells |
| TNFR1 | TNF receptor 1 |
| TNF-α | Tumor necrosis factor alpha |
| uPAR | Urokinase-type plasminogen activator receptor |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cell |
| X-Gal | X-galactosidase |
| α-SMA | α-smooth muscle actin |
| γ-H2AX | Phosphorylated H2A histone family member X |
References
- Ageing and Health. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 20 March 2023).
- Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 20 March 2023).
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. 2019, 18, e12931. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Kim, S.R.; Jiang, K.; Ogrodnik, M.; Zhu, X.Y.; Ferguson, C.M.; Tchkonia, T.; Lerman, A.; Kirkland, J.L.; Lerman, L.O. Quercetin Reverses Cardiac Systolic Dysfunction in Mice Fed with a High-Fat Diet: Role of Angiogenesis. Oxid. Med. Cell. Longev. 2021, 2021, 8875729. [Google Scholar] [CrossRef]
- Gunasekaran, U.; Gannon, M. Type 2 diabetes and the aging pancreatic beta cell. Aging 2011, 3, 565–575. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; Kirkland, J.L. Targeting cellular senescence in metabolic disease. Mol. Metab. 2022, 66, 101601. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Gasek, N.S.; Zhou, Y.; Cohn, R.L.; Martin, D.E.; Zuo, W.; Flynn, W.F.; Guo, C.; Jellison, E.R.; et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 2022, 34, 75–89.e78. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Obesity, Senescence, and Senolytics. Handb. Exp. Pharmacol. 2022, 274, 165–180. [Google Scholar] [CrossRef]
- Conley, S.M.; Hickson, L.J.; Kellogg, T.A.; McKenzie, T.; Heimbach, J.K.; Taner, T.; Tang, H.; Jordan, K.L.; Saadiq, I.M.; Woollard, J.R.; et al. Human Obesity Induces Dysfunction and Early Senescence in Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells. Front. Cell Dev. Biol. 2020, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 2015, 4, e12997. [Google Scholar] [CrossRef] [PubMed]
- Escande, C.; Nin, V.; Pirtskhalava, T.; Chini, C.C.; Thereza Barbosa, M.; Mathison, A.; Urrutia, R.; Tchkonia, T.; Kirkland, J.L.; Chini, E.N. Deleted in Breast Cancer 1 regulates cellular senescence during obesity. Aging Cell 2014, 13, 951–953. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Meijnikman, A.S.; van Olden, C.C.; Aydin, Ö.; Herrema, H.; Kaminska, D.; Lappa, D.; Männistö, V.; Tremaroli, V.; Olofsson, L.E.; de Brauw, M.; et al. Hyperinsulinemia Is Highly Associated With Markers of Hepatocytic Senescence in Two Independent Cohorts. Diabetes 2022, 71, 1929–1936. [Google Scholar] [CrossRef]
- Baboota, R.K.; Rawshani, A.; Bonnet, L.; Li, X.; Yang, H.; Mardinoglu, A.; Tchkonia, T.; Kirkland, J.L.; Hoffmann, A.; Dietrich, A.; et al. BMP4 and Gremlin 1 regulate hepatic cell senescence during clinical progression of NAFLD/NASH. Nat. Metab. 2022, 4, 1007–1021. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Nalysnyk, L.; Cid-Ruzafa, J.; Rotella, P.; Esser, D. Incidence and prevalence of idiopathic pulmonary fibrosis: Review of the literature. Eur. Respir. Rev. 2012, 21, 355–361. [Google Scholar] [CrossRef]
- Parikh, P.; Britt, R.D., Jr.; Manlove, L.J.; Wicher, S.A.; Roesler, A.; Ravix, J.; Teske, J.; Thompson, M.A.; Sieck, G.C.; Kirkland, J.L.; et al. Hyperoxia-induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 51–60. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Griffin, T.P.; Zhu, X.; Islam, M.N.; Conley, S.M.; Eirin, A.; Tang, H.; O’Shea, P.M.; Palmer, A.K.; McCoy, R.G.; et al. Senescence marker activin A is increased in human diabetic kidney disease: Association with kidney function and potential implications for therapy. BMJ Open. Diabetes Res. Care 2019, 7, e000720. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Jiang, K.; Ogrodnik, M.; Chen, X.; Zhu, X.Y.; Lohmeier, H.; Ahmed, L.; Tang, H.; Tchkonia, T.; Hickson, L.J.; et al. Increased renal cellular senescence in murine high-fat diet: Effect of the senolytic drug quercetin. Transl. Res. 2019, 213, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Rowsey, J.L.; Eckhardt, B.A.; Thicke, B.S.; Fraser, D.G.; Tchkonia, T.; Kirkland, J.L.; Monroe, D.G.; Khosla, S. Independent Roles of Estrogen Deficiency and Cellular Senescence in the Pathogenesis of Osteoporosis: Evidence in Young Adult Mice and Older Humans. J. Bone Miner. Res. 2019, 34, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Farr, J.N.; Kirkland, J.L. Inhibiting Cellular Senescence: A New Therapeutic Paradigm for Age-Related Osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Monroe, D.G.; Park, S.; Hachfeld, C.; Tchkonia, T.; Kirkland, J.L.; Khosla, S.; Passos, J.F.; et al. Targeted Reduction of Senescent Cell Burden Alleviates Focal Radiotherapy-Related Bone Loss. J. Bone Miner. Res. 2020, 35, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Doolittle, M.; Tchkonia, T.; Kirkland, J.L.; LeBrasseur, N.K.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 2022, 21, e13602. [Google Scholar] [CrossRef]
- Cavalcante, M.B.; Saccon, T.D.; Nunes, A.D.C.; Kirkland, J.L.; Tchkonia, T.; Schneider, A.; Masternak, M.M. Dasatinib plus quercetin prevents uterine age-related dysfunction and fibrosis in mice. Aging 2020, 12, 2711–2722. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077.e1068. [Google Scholar] [CrossRef]
- de Magalhães, J.P. How ageing processes influence cancer. Nat. Rev. Cancer 2013, 13, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Suda, M.; Shimizu, I.; Yoshida, Y.; Katsuumi, G.; Minamino, T. Chapter 23—Endothelial cell dysfunction and senescence: Biologic mechanisms and hemodynamic consequences. In Textbook of Arterial Stiffness and Pulsatile Hemodynamics in Health and Disease; Chirinos, J.A., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 359–367. [Google Scholar]
- Hwang, H.J.; Kim, N.; Herman, A.B.; Gorospe, M.; Lee, J.S. Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. Int. J. Mol. Sci. 2022, 23, 10135. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Hayashi, Y.; Suda, M.; Tateno, K.; Okada, S.; Moriya, J.; Yokoyama, M.; Nojima, A.; Yamashita, M.; Kobayashi, Y.; et al. Notch Signaling Regulates the Lifespan of Vascular Endothelial Cells via a p16-Dependent Pathway. PLoS ONE 2014, 9, e100359. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J.; Kim, S.H.; Lim, C.S.; Rubio, M. Cellular senescence, cancer and aging: The telomere connection. Exp. Gerontol. 2001, 36, 1619–1637. [Google Scholar] [CrossRef]
- Shimizu, I.; Yoshida, Y.; Suda, M.; Minamino, T. DNA damage response and metabolic disease. Cell Metab. 2014, 20, 967–977. [Google Scholar] [CrossRef]
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e327. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef]
- Miyauchi, H.; Minamino, T.; Tateno, K.; Kunieda, T.; Toko, H.; Komuro, I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004, 23, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett. 2001, 493, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J. Cell. Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M. SASP reflects senescence. EMBO Rep. 2009, 10, 228–230. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. Embo J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. Embo J. 2019, 38, e100492. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.G.; Tang, Q.Z. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis. 2022, 13, 103–128. [Google Scholar] [CrossRef]
- Conover, C.A.; Bale, L.K. Senescence induces proteolytically-active PAPP-A secretion and association with extracellular vesicles in human pre-adipocytes. Exp. Gerontol. 2022, 172, 112070. [Google Scholar] [CrossRef] [PubMed]
- Conover, C.A. Key questions and answers about pregnancy-associated plasma protein-A. Trends Endocrinol. Metab. 2012, 23, 242–249. [Google Scholar] [CrossRef]
- Conover, C.A.; Bale, L.K.; Marler, R.J. Pregnancy-associated plasma protein-A deficiency improves survival of mice on a high fat diet. Exp. Gerontol. 2015, 70, 131–134. [Google Scholar] [CrossRef]
- Conover, C.A.; Harstad, S.L.; Tchkonia, T.; Kirkland, J.L. Preferential impact of pregnancy-associated plasma protein-A deficiency on visceral fat in mice on high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1145–E1153. [Google Scholar] [CrossRef]
- Bale, L.K.; West, S.A.; Conover, C.A. Inducible knockdown of pregnancy-associated plasma protein-A gene expression in adult female mice extends life span. Aging Cell 2017, 16, 895–897. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e2654. [Google Scholar] [CrossRef]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell. Rep. 2021, 36, 109441. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, U.; Misra, A.; Tchkonia, T.; Kirkland, J.L. Impact of Senescent Cell Subtypes on Tissue Dysfunction and Repair: Importance and Research Questions. Mech. Ageing Dev. 2021, 198, 111548. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Saul, D.; Kosinsky, R.L.; Atkinson, E.J.; Doolittle, M.L.; Zhang, X.; LeBrasseur, N.K.; Pignolo, R.J.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 2022, 13, 4827. [Google Scholar] [CrossRef]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef]
- Englund, D.A.; Sakamoto, A.E.; Fritsche, C.M.; Heeren, A.A.; Zhang, X.; Kotajarvi, B.R.; Lecy, D.R.; Yousefzadeh, M.J.; Schafer, M.J.; White, T.A.; et al. Exercise reduces circulating biomarkers of cellular senescence in humans. Aging Cell 2021, 20, e13415. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Prata, L.; Gerdes, E.O.W.; Netto, J.M.E.; Pirtskhalava, T.; Giorgadze, N.; Tripathi, U.; Inman, C.L.; Johnson, K.O.; Xue, A.; et al. Orally-active, clinically-translatable senolytics restore α-Klotho in mice and humans. EBioMedicine 2022, 77, 103912. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Takahashi, A. Senescence-associated extracellular vesicle release plays a role in senescence-associated secretory phenotype (SASP) in age-associated diseases. J. Biochem. 2021, 169, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Hitomi, K.; Miyata, K.; Tanaka, Y.; Fujii, R.; Chiba, M.; Loo, T.M.; Hanyu, A.; Kawasaki, H.; Kato, H.; et al. Identification of Novel Senescent Markers in Small Extracellular Vesicles. Int. J. Mol. Sci. 2023, 24, 2421. [Google Scholar] [CrossRef]
- Munk, R.; Panda, A.C.; Grammatikakis, I.; Gorospe, M.; Abdelmohsen, K. Senescence-Associated MicroRNAs. Int. Rev. Cell Mol. Biol. 2017, 334, 177–205. [Google Scholar] [CrossRef]
- Suh, N. MicroRNA controls of cellular senescence. BMB Rep. 2018, 51, 493–499. [Google Scholar] [CrossRef]
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973. [Google Scholar] [CrossRef]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 2017, 541, 96–101. [Google Scholar] [CrossRef]
- Sato, Y.; Oguchi, A.; Fukushima, Y.; Masuda, K.; Toriu, N.; Taniguchi, K.; Yoshikawa, T.; Cui, X.; Kondo, M.; Hosoi, T.; et al. CD153/CD30 signaling promotes age-dependent tertiary lymphoid tissue expansion and kidney injury. Journal. Clin. Investig. 2022, 132, e146071. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.D.; Bulavin, D.V. Defined p16(High) Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e86. [Google Scholar] [CrossRef]
- Toya, T.; Ahmad, A.; Attia, Z.; Cohen-Shelly, M.; Ozcan, I.; Noseworthy, P.A.; Lopez-Jimenez, F.; Kapa, S.; Lerman, L.O.; Friedman, P.A.; et al. Vascular Aging Detected by Peripheral Endothelial Dysfunction Is Associated With ECG-Derived Physiological Aging. J. Am. Heart Assoc. 2021, 10, e018656. [Google Scholar] [CrossRef] [PubMed]
- Kotla, S.; Vu, H.T.; Ko, K.A.; Wang, Y.; Imanishi, M.; Heo, K.S.; Fujii, Y.; Thomas, T.N.; Gi, Y.J.; Mazhar, H.; et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight 2019, 4, e124867. [Google Scholar] [CrossRef] [PubMed]
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef]
- Liu, J.; Chen, S.; Biswas, S.; Nagrani, N.; Chu, Y.; Chakrabarti, S.; Feng, B. Glucose-induced oxidative stress and accelerated aging in endothelial cells are mediated by the depletion of mitochondrial SIRTs. Physiol. Rep. 2020, 8, e14331. [Google Scholar] [CrossRef]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; VanDongen, N.S.; D’Alessandro, A.; Reisz, J.A.; Ziemba, B.P.; Seals, D.R. Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. Cardio Oncol. 2020, 2, 475–488. [Google Scholar] [CrossRef]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; Casso, A.G.; Ziemba, B.P.; Melov, S.; Campisi, J.; Seals, D.R. Tumor Necrosis Factor Alpha-Mediated Inflammation and Remodeling of the Extracellular Matrix Underlies Aortic Stiffening Induced by the Common Chemotherapeutic Agent Doxorubicin. Hypertension 2021, 77, 1581–1590. [Google Scholar] [CrossRef]
- Dagher, O.; Mury, P.; Noly, P.E.; Fortier, A.; Lettre, G.; Thorin, E.; Carrier, M. Design of a Randomized Placebo-Controlled Trial to Evaluate the Anti-inflammatory and Senolytic Effects of Quercetin in Patients Undergoing Coronary Artery Bypass Graft Surgery. Front. Cardiovasc. Med. 2021, 8, 741542. [Google Scholar] [CrossRef]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Shimizu, I.; Nagasawa, A.; Yoshida, Y.; Katsuumi, G.; Wakasugi, T.; Hayashi, Y.; Ikegami, R.; Suda, M.; Ota, Y.; et al. p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J. Mol. Cell. Cardiol. 2019, 129, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Okada, S.; Nakagomi, A.; Moriya, J.; Shimizu, I.; Nojima, A.; Yoshida, Y.; Ichimiya, H.; Kamimura, N.; Kobayashi, Y.; et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep. 2014, 7, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef]
- Baumgartner-Parzer, S.M.; Waldhäusl, W.K. The endothelium as a metabolic and endocrine organ: Its relation with insulin resistance. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. 2), S166–S179. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.R.Y.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ. Heart Fail. 2017, 10, e003806. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef]
- Garfinkel, S.; Brown, S.; Wessendorf, J.H.; Maciag, T. Post-transcriptional regulation of interleukin 1 alpha in various strains of young and senescent human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Matacchione, G.; Gurău, F.; Silvestrini, A.; Tiboni, M.; Mancini, L.; Valli, D.; Rippo, M.R.; Recchioni, R.; Marcheselli, F.; Carnevali, O.; et al. Anti-SASP and anti-inflammatory activity of resveratrol, curcumin and β-caryophyllene association on human endothelial and monocytic cells. Biogerontology 2021, 22, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: Current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 2023, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- van der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 2020, 12, eaaw4974. [Google Scholar] [CrossRef] [PubMed]
- Culley, M.K.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Perk, D.; Negi, V.; Yu, Q.; Woodcock, C.C.; Handen, A.; Speyer, G.; et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J. Clin. Investig. 2021, 131, e136459. [Google Scholar] [CrossRef]
- Chang, H.; Rha, S.Y.; Jeung, H.C.; Park, K.H.; Kim, T.S.; Kim, Y.B.; Chung, H.C. Telomerase- and angiogenesis-related gene responses to irradiation in human umbilical vein endothelial cells. Int. J. Mol. Med. 2013, 31, 1202–1208. [Google Scholar] [CrossRef]
- Suda, M.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Katsuumi, G.; Wakasugi, T.; Yoshida, Y.; Okuda, S.; Soga, T.; et al. Inhibition of dipeptidyl peptidase-4 ameliorates cardiac ischemia and systolic dysfunction by up-regulating the FGF-2/EGR-1 pathway. PLoS ONE 2017, 12, e0182422. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Chi, C.; Li, D.J.; Jiang, Y.J.; Tong, J.; Fu, H.; Wu, Y.H.; Shen, F.M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef]
- Bennett, M.R.; Macdonald, K.; Chan, S.W.; Boyle, J.J.; Weissberg, P.L. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ. Res. 1998, 82, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.P.; Bennett, M.R. The role of mitochondrial DNA damage in the development of atherosclerosis. Free. Radic. Biol. Med. 2016, 100, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288. [Google Scholar] [CrossRef] [PubMed]
- Otuki, S.; Izumi, D.; Suda, M.; Sato, A.; Hasegawa, Y.; Yagihara, N.; Iijima, K.; Chinushi, M.; Fuse, I.; Minamino, T. Effects of Direct Oral Anticoagulants at the Peak Phase, Trough Phase, and After Vascular Injury. J. Am. Coll. Cardiol. 2018, 71, 102–104. [Google Scholar] [CrossRef]
- Hayashi, Y.; Shimizu, I.; Yoshida, Y.; Ikegami, R.; Suda, M.; Katsuumi, G.; Fujiki, S.; Ozaki, K.; Abe, M.; Sakimura, K.; et al. Coagulation factors promote brown adipose tissue dysfunction and abnormal systemic metabolism in obesity. iScience 2022, 25, 104547. [Google Scholar] [CrossRef]
- Sanada, F.; Muratsu, J.; Otsu, R.; Shimizu, H.; Koibuchi, N.; Uchida, K.; Taniyama, Y.; Yoshimura, S.; Rakugi, H.; Morishita, R. Local Production of Activated Factor X in Atherosclerotic Plaque Induced Vascular Smooth Muscle Cell Senescence. Sci. Rep. 2017, 7, 17172. [Google Scholar] [CrossRef]
- Bennett, M.R.; Clarke, M.C. Basic research: Killing the old: Cell senescence in atherosclerosis. Nat. Rev. Cardiol. 2016, 14, 8–9. [Google Scholar] [CrossRef]
- Furuuchi, R.; Shimizu, I.; Yoshida, Y.; Katsuumi, G.; Suda, M.; Kubota, Y.; Walsh, K.; Minamino, T. Endothelial SIRT-1 has a critical role in the maintenance of capillarization in brown adipose tissue. iScience 2022, 25, 105424. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Finigan, A.; Figg, N.L.; Uryga, A.K.; Bennett, M.R. SIRT6 Protects Smooth Muscle Cells From Senescence and Reduces Atherosclerosis. Circ. Res. 2021, 128, 474–491. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef]
- Rubio-Ruiz, M.E.; Pérez-Torres, I.; Soto, M.E.; Pastelín, G.; Guarner-Lans, V. Aging in blood vessels. Medicinal agents FOR systemic arterial hypertension in the elderly. Ageing Res. Rev. 2014, 18, 132–147. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Childs, B.G.; Zhang, C.; Shuja, F.; Sturmlechner, I.; Trewartha, S.; Velasco, R.F.; Baker, D.J.; Li, H.; van Deursen, J.M. Senescent cells suppress innate smooth muscle cell repair functions in atherosclerosis. Nat. Aging 2021, 1, 698–714. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovasc. Res. 2018, 114, 513–528. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef]
- Nakano-Kurimoto, R.; Ikeda, K.; Uraoka, M.; Nakagawa, Y.; Yutaka, K.; Koide, M.; Takahashi, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1673–H1684. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Luo, M.; Yan, D.; Zhang, C. Carnosine attenuates vascular smooth muscle cells calcification through mTOR signaling pathway. Aging Med. 2020, 3, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Suda, M.; Katsuumi, G.; Nakao, M.; Hoyano, M.; Kashimura, T.; et al. Role of smooth muscle cell p53 in pulmonary arterial hypertension. PLoS ONE 2019, 14, e0212889. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Ball, A.J.; Levine, F. Telomere-independent cellular senescence in human fetal cardiomyocytes. Aging Cell 2005, 4, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, C380–C391. [Google Scholar] [CrossRef]
- Zhang, F.-X.; Chen, M.-L.; Shan, Q.-J.; Zou, J.-G.; Chen, C.; Yang, B.; Xu, D.-J.; Jin, Y.; Cao, K.-J. Hypoxia reoxygenation induces premature senescence in neonatal SD rat cardiomyocytes. Acta Pharmacol. Sin. 2007, 28, 44–51. [Google Scholar] [CrossRef]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef] [PubMed]
- Spallarossa, P.; Altieri, P.; Aloi, C.; Garibaldi, S.; Barisione, C.; Ghigliotti, G.; Fugazza, G.; Barsotti, A.; Brunelli, C. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2169–H2181. [Google Scholar] [CrossRef]
- Maejima, Y.; Adachi, S.; Ito, H.; Hirao, K.; Isobe, M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell 2008, 7, 125–136. [Google Scholar] [CrossRef]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.-L. Nitric Oxide and Cardiac Function. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef]
- Shimizu, I.; Yoshida, Y.; Katsuno, T.; Tateno, K.; Okada, S.; Moriya, J.; Yokoyama, M.; Nojima, A.; Ito, T.; Zechner, R.; et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012, 15, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Xue, L.; Yang, F.; Dai, S.; Han, Z.; Liu, K.; Liu, B.; Yuan, Q.; Cui, Z.; Zhang, Y.; et al. Postinfarction Hearts Are Protected by Premature Senescent Cardiomyocytes Via GATA 4-Dependent CCN 1 Secretion. J. Am. Heart Assoc. 2018, 7, e009111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, C.; Liu, C.; Wang, Y.; Zhang, W.; Xing, Y. Trimetazidine and l-carnitine prevent heart aging and cardiac metabolic impairment in rats via regulating cardiac metabolic substrates. Exp. Gerontol. 2019, 119, 120–127. [Google Scholar] [CrossRef]
- Dillon, L.M.; Rebelo, A.P.; Moraes, C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012, 64, 231–241. [Google Scholar] [CrossRef]
- Gélinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef]
- Turdi, S.; Fan, X.; Li, J.; Zhao, J.; Huff, A.F.; Du, M.; Ren, J. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 2010, 9, 592–606. [Google Scholar] [CrossRef]
- Tang, X.; Chen, X.F.; Wang, N.Y.; Wang, X.M.; Liang, S.T.; Zheng, W.; Lu, Y.B.; Zhao, X.; Hao, D.L.; Zhang, Z.Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Burke, A.P.; Farb, A.; Malcom, G.T.; Liang, Y.H.; Smialek, J.; Virmani, R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med. 1997, 336, 1276–1282. [Google Scholar] [CrossRef]
- Libby, P.; Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Diversity of Denizens of the Atherosclerotic Plaque. Circulation 2008, 117, 3168–3170. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C.; George, S.J.; Ismail, Y.; Johnson, J.L.; Sala-Newby, G.B.; Thomas, A.C. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb. Haemost. 2009, 101, 1006–1011. [Google Scholar] [PubMed]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abeßer, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e520. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e123. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Richardson, G.D.; Sage, A.; Bennaceur, K.; Al Zhrany, N.; Coelho-Lima, J.; Dookun, E.; Draganova, L.; Saretzki, G.; Breault, D.T.; Mallat, Z.; et al. Telomerase Mediates Lymphocyte Proliferation but Not the Atherosclerosis-Suppressive Potential of Regulatory T-Cells. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Bernal, E.; Martinez, M.; Torres, A.; Guillamón, C.F.; Alcaraz, A.; Alcaraz, M.J.; Muñoz, A.; Valero, S.; Botella, C.; Campillo, J.A.; et al. T cell senescence predicts subclinical atherosclerosis in HIV-infected patients similarly to traditional cardiovascular risk factors. Antiviral Res. 2019, 162, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.T.; Park, S.; Shin, E.C.; Lee, W.W. T cell senescence and cardiovascular diseases. Clin. Exp. Med. 2016, 16, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Brouilette, S.W.; Whittaker, A.; Stevens, S.E.; van der Harst, P.; Goodall, A.H.; Samani, N.J. Telomere length is shorter in healthy offspring of subjects with coronary artery disease: Support for the telomere hypothesis. Heart 2008, 94, 422–425. [Google Scholar] [CrossRef]
- Varricchi, G.; Bencivenga, L.; Poto, R.; Pecoraro, A.; Shamji, M.H.; Rengo, G. The emerging role of T follicular helper (T(FH)) cells in aging: Influence on the immune frailty. Ageing Res. Rev. 2020, 61, 101071. [Google Scholar] [CrossRef]
- Bazioti, V.; La Rose, A.M.; Maassen, S.; Bianchi, F.; de Boer, R.; Halmos, B.; Dabral, D.; Guilbaud, E.; Flohr-Svendsen, A.; Groenen, A.G.; et al. T cell cholesterol efflux suppresses apoptosis and senescence and increases atherosclerosis in middle aged mice. Nat. Commun. 2022, 13, 3799. [Google Scholar] [CrossRef]
- Youn, J.C.; Yu, H.T.; Lim, B.J.; Koh, M.J.; Lee, J.; Chang, D.Y.; Choi, Y.S.; Lee, S.H.; Kang, S.M.; Jang, Y.; et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 2013, 62, 126–133. [Google Scholar] [CrossRef]
- Tae Yu, H.; Youn, J.C.; Lee, J.; Park, S.; Chi, H.S.; Lee, J.; Choi, C.; Park, S.; Choi, D.; Ha, J.W.; et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell. Mol. Immunol. 2015, 12, 466–473. [Google Scholar] [CrossRef]
- Kaplan, R.C.; Sinclair, E.; Landay, A.L.; Lurain, N.; Sharrett, A.R.; Gange, S.J.; Xue, X.; Hunt, P.; Karim, R.; Kern, D.M.; et al. T cell activation and senescence predict subclinical carotid artery disease in HIV-infected women. J. Infect. Dis. 2011, 203, 452–463. [Google Scholar] [CrossRef]
- Liuzzo, G.; Biasucci, L.M.; Trotta, G.; Brugaletta, S.; Pinnelli, M.; Digianuario, G.; Rizzello, V.; Rebuzzi, A.G.; Rumi, C.; Maseri, A.; et al. Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J. Am. Coll. Cardiol. 2007, 50, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Delgobo, M.; Heinrichs, M.; Hapke, N.; Ashour, D.; Appel, M.; Srivastava, M.; Heckel, T.; Spyridopoulos, I.; Hofmann, U.; Frantz, S.; et al. Terminally Differentiated CD4(+) T Cells Promote Myocardial Inflammaging. Front. Immunol. 2021, 12, 584538. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kalka, C.; Masuda, H.; Chen, D.; Silver, M.; Kearney, M.; Magner, M.; Isner, J.M.; Asahara, T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med. 1999, 5, 434–438. [Google Scholar] [CrossRef]
- Werner, N.; Nickenig, G. Clinical and therapeutical implications of EPC biology in atherosclerosis. J. Cell. Mol. Med. 2006, 10, 318–332. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, E1–E7. [Google Scholar] [CrossRef]
- Yang, J.; Chang, E.; Cherry, A.M.; Bangs, C.D.; Oei, Y.; Bodnar, A.; Bronstein, A.; Chiu, C.P.; Herron, G.S. Human endothelial cell life extension by telomerase expression. J. Biol. Chem. 1999, 274, 26141–26148. [Google Scholar] [CrossRef]
- Torella, D.; Rota, M.; Nurzynska, D.; Musso, E.; Monsen, A.; Shiraishi, I.; Zias, E.; Walsh, K.; Rosenzweig, A.; Sussman, M.A.; et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ. Res. 2004, 94, 514–524. [Google Scholar] [CrossRef]
- Cesselli, D.; Beltrami, A.P.; D’Aurizio, F.; Marcon, P.; Bergamin, N.; Toffoletto, B.; Pandolfi, M.; Puppato, E.; Marino, L.; Signore, S.; et al. Effects of age and heart failure on human cardiac stem cell function. Am. J. Pathol. 2011, 179, 349–366. [Google Scholar] [CrossRef]
- Castaldi, A.; Dodia, R.M.; Orogo, A.M.; Zambrano, C.M.; Najor, R.H.; Gustafsson, Å.B.; Heller Brown, J.; Purcell, N.H. Decline in cellular function of aged mouse c-kit(+) cardiac progenitor cells. J. Physiol. 2017, 595, 6249–6262. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.; Scalise, M.; Salerno, N.; Salerno, L.; Molinaro, C.; Cappetta, D.; Torella, M.; Greco, M.; Foti, D.; Sasso, F.C.; et al. Diabetes-Induced Cellular Senescence and Senescence-Associated Secretory Phenotype Impair Cardiac Regeneration and Function Independently of Age. Diabetes 2022, 71, 1081–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.H.; Du, J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhang, W.; Ma, Y.; Chen, B.; Liu, Y.; Piao, C.; Wang, Y.; Yang, M.; Liu, T.; Zhang, J.; et al. Haplodeficiency of Ataxia Telangiectasia Mutated Accelerates Heart Failure After Myocardial Infarction. J. Am. Heart Assoc. 2017, 6, e006349. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef]
- Li, K.; Li, Y.; Yu, Y.; Ding, J.; Huang, H.; Chu, C.; Hu, L.; Yu, Y.; Cao, Y.; Xu, P.; et al. Bmi-1 alleviates adventitial fibroblast senescence by eliminating ROS in pulmonary hypertension. BMC Pulm. Med. 2021, 21, 80. [Google Scholar] [CrossRef]
- Roger, I.; Milara, J.; Belhadj, N.; Cortijo, J. Senescence Alterations in Pulmonary Hypertension. Cells 2021, 10, 3456. [Google Scholar] [CrossRef]
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P.; et al. Visceral Adipose Tissue Drives Cardiac Aging Through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822. [Google Scholar] [CrossRef]
- Kamo, T.; Akazawa, H.; Komuro, I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ. Res. 2015, 117, 89–98. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef]
- Civitarese, R.A.; Kapus, A.; McCulloch, C.A.; Connelly, K.A. Role of integrins in mediating cardiac fibroblast-cardiomyocyte cross talk: A dynamic relationship in cardiac biology and pathophysiology. Basic. Res. Cardiol. 2017, 112, 6. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, M.; Higo, T.; Naito, A.T.; Nakagawa, A.; Sumida, T.; Okada, K.; Sakai, T.; Kuramoto, Y.; Yamaguchi, T.; Ito, M.; et al. Activation of DNA Damage Response and Cellular Senescence in Cardiac Fibroblasts Limit Cardiac Fibrosis After Myocardial Infarction. Int. Heart J. 2019, 60, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- The Impact of Chronic Disease on Aging Americans. Available online: https://www.cvshealth.com/news-and-insights/articles/by-the-numbers-the-impact-of-chronic-disease-on-aging-americans#:~:text=According%20to%20the%20National%20Council,Hypertension%20(46%20percent) (accessed on 20 March 2023).
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, U.; Nchioua, R.; Prata, L.; Zhu, Y.; Gerdes, E.O.W.; Giorgadze, N.; Pirtskhalava, T.; Parker, E.; Xue, A.; Espindola-Netto, J.M.; et al. SARS-CoV-2 causes senescence in human cells and exacerbates the senescence-associated secretory phenotype through TLR-3. Aging 2021, 13, 21838–21854. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Chang, Q.; Jorgensen, C.; Pawson, T.; Hedley, D.W. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br. J. Cancer 2008, 99, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.Q.; Wu, X.S.; Wei, B.; Chen, L. Eph receptors and ephrins as targets for cancer therapy. J. Cell. Mol. Med. 2012, 16, 2894–2909. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Palmer, A.K.; Kirkland, J.L. New Horizons: Novel Approaches to Enhance Healthspan Through Targeting Cellular Senescence and Related Aging Mechanisms. J. Clin. Endocrinol. Metab. 2021, 106, e1481–e1487. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Xu, Q.; Fu, Q.; Li, Z.; Liu, H.; Wang, Y.; Lin, X.; He, R.; Zhang, X.; Ju, Z.; Campisi, J.; et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat. Metab. 2021, 3, 1706–1726. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e116. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N. Engl. J. Med. 1997, 336, 525–533. [Google Scholar] [CrossRef]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martínez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Hsiao, C.L.; Yoshida, Y.; Matsumoto, N.; Yoshida, Y.; Katayama, A.; Wada, J.; Seki, M.; et al. Glycoprotein nonmetastatic melanoma protein B regulates lysosomal integrity and lifespan of senescent cells. Sci. Rep. 2022, 12, 6522. [Google Scholar] [CrossRef]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Matsumoto, N.; Yoshida, Y.; Mikawa, R.; Katayama, A.; et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat. Aging 2021, 1, 1117–1126. [Google Scholar] [CrossRef]
- Poblocka, M.; Bassey, A.L.; Smith, V.M.; Falcicchio, M.; Manso, A.S.; Althubiti, M.; Sheng, X.; Kyle, A.; Barber, R.; Frigerio, M.; et al. Targeted clearance of senescent cells using an antibody-drug conjugate against a specific membrane marker. Sci. Rep. 2021, 11, 20358. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Martín-Caballero, J.; Flores, J.M.; García-Palencia, P.; Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001, 61, 6234–6238. [Google Scholar] [PubMed]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes. Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef] [PubMed]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Cañamero, M.; Rayon, T.; Ors, I.; Graña, O.; Megías, D.; Domínguez, O.; Martínez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e1712. [Google Scholar] [CrossRef]
- Browder, K.C.; Reddy, P.; Yamamoto, M.; Haghani, A.; Guillen, I.G.; Sahu, S.; Wang, C.; Luque, Y.; Prieto, J.; Shi, L.; et al. In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat. Aging 2022, 2, 243–253. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Huffman, D.M.; Justice, J.N.; Stout, M.B.; Kirkland, J.L.; Barzilai, N.; Austad, S.N. Evaluating Health Span in Preclinical Models of Aging and Disease: Guidelines, Challenges, and Opportunities for Geroscience. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1395–1406. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Bannister, C.A.; Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Halcox, J.P.; Schernthaner, G.; Mukherjee, J.; Currie, C.J. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes. Metab. 2014, 16, 1165–1173. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, X.; Xie, W.; Han, P.; Li, K.; Sun, Z.; Wang, Y.; Chen, C.; Song, R.; Cao, C.; et al. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart. Circ. Res. 2010, 106, 317–327. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Hurez, V.; Dao, V.; Liu, A.; Pandeswara, S.; Gelfond, J.; Sun, L.; Bergman, M.; Orihuela, C.J.; Galvan, V.; Padrón, Á.; et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015, 14, 945–956. [Google Scholar] [CrossRef]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef]
- Walters, H.E.; Cox, L.S. mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2325. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef]
- Targeting the Biology of Aging. Ushering a New Era of Interventions. Available online: https://www.afar.org/tame-trial (accessed on 20 March 2023).
- Justice, J.N.; Ferrucci, L.; Newman, A.B.; Aroda, V.R.; Bahnson, J.L.; Divers, J.; Espeland, M.A.; Marcovina, S.; Pollak, M.N.; Kritchevsky, S.B.; et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: Report from the TAME Biomarkers Workgroup. Geroscience 2018, 40, 419–436. [Google Scholar] [CrossRef]
- Sunderland, P.; Alshammari, L.; Ambrose, E.; Torella, D.; Ellison-Hughes, G.M. Senolytics rejuvenate the reparative activity of human cardiomyocytes and endothelial cells. J. Cardiovasc. Aging 2023, 3, 21. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Panchal, S.K.; Poudyal, H.; Brown, L. Quercetin ameliorates cardiovascular, hepatic, and metabolic changes in diet-induced metabolic syndrome in rats. J. Nutr. 2012, 142, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Lérida-Viso, A.; Estepa-Fernández, A.; Morellá-Aucejo, Á.; Lozano-Torres, B.; Alfonso, M.; Blandez, J.F.; Bisbal, V.; Sepúlveda, P.; García-Fernández, A.; Orzáez, M.; et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 2022, 183, 106356. [Google Scholar] [CrossRef]
- Iske, J.; Seyda, M.; Heinbokel, T.; Maenosono, R.; Minami, K.; Nian, Y.; Quante, M.; Falk, C.S.; Azuma, H.; Martin, F.; et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat. Commun. 2020, 11, 4289. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Dai, Y.; Liu, A.; Li, X.; Wu, L.; Lu, L.; Bao, Y.; Jin, Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J. Cardiovasc. Pharmacol. 2020, 76, 452–460. [Google Scholar] [CrossRef]
- Xintarakou, A.; Tzeis, S.; Psarras, S.; Asvestas, D.; Vardas, P. Atrial fibrosis as a dominant factor for the development of atrial fibrillation: Facts and gaps. Europace 2020, 22, 342–351. [Google Scholar] [CrossRef]
- Platonov, P.G.; Mitrofanova, L.B.; Orshanskaya, V.; Ho, S.Y. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J. Am. Coll. Cardiol. 2011, 58, 2225–2232. [Google Scholar] [CrossRef]
- Adili, A.; Zhu, X.; Cao, H.; Tang, X.; Wang, Y.; Wang, J.; Shi, J.; Zhou, Q.; Wang, D. Atrial Fibrillation Underlies Cardiomyocyte Senescence and Contributes to Deleterious Atrial Remodeling during Disease Progression. Aging Dis. 2022, 13, 298–312. [Google Scholar] [CrossRef]
- Jesel, L.; Abbas, M.; Park, S.H.; Matsushita, K.; Kindo, M.; Hasan, H.; Auger, C.; Sato, C.; Ohlmann, P.; Mazzucotelli, J.P.; et al. Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression. J. Clin. Med. 2019, 9, 36. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, W.; Hu, Y.; Wan, Z.; Bai, H.; Yang, Q.; Zheng, Q. Quercetin improves atrial fibrillation through inhibiting TGF-β/Smads pathway via promoting MiR-135b expression. Phytomedicine 2021, 93, 153774. [Google Scholar] [CrossRef]
- Liu, L.; Gan, S.; Li, B.; Ge, X.; Yu, H.; Zhou, H. Fisetin Alleviates Atrial Inflammation, Remodeling, and Vulnerability to Atrial Fibrillation after Myocardial Infarction. Int. Heart J. 2019, 60, 1398–1406. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Hu, C.; Pan, Q.; Wang, B.; Li, X.; Geng, J.; Xu, B. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget 2017, 8, 57981–57990. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Park, B.W.; Park, J.H.; Lim, S.; Kwon, S.P.; Hwang, J.W.; Kim, H.; Park, H.J.; Kim, B.S. Local delivery of a senolytic drug in ischemia and reperfusion-injured heart attenuates cardiac remodeling and restores impaired cardiac function. Acta Biomater. 2021, 135, 520–533. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [CrossRef]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 2020, 19, e13249. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef] [PubMed]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marbán, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef]
- Hare, J.M.; DiFede, D.L.; Rieger, A.C.; Florea, V.; Landin, A.M.; El-Khorazaty, J.; Khan, A.; Mushtaq, M.; Lowery, M.H.; Byrnes, J.J.; et al. Randomized Comparison of Allogeneic Versus Autologous Mesenchymal Stem Cells for Nonischemic Dilated Cardiomyopathy: POSEIDON-DCM Trial. J. Am. Coll. Cardiol. 2017, 69, 526–537. [Google Scholar] [CrossRef]
- Alexandre, J.; Cautela, J.; Ederhy, S.; Damaj, G.L.; Salem, J.E.; Barlesi, F.; Farnault, L.; Charbonnier, A.; Mirabel, M.; Champiat, S.; et al. Cardiovascular Toxicity Related to Cancer Treatment: A Pragmatic Approach to the American and European Cardio-Oncology Guidelines. J. Am. Heart Assoc. 2020, 9, e018403. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- Bulten, B.F.; Sollini, M.; Boni, R.; Massri, K.; de Geus-Oei, L.F.; van Laarhoven, H.W.M.; Slart, R.; Erba, P.A. Cardiac molecular pathways influenced by doxorubicin treatment in mice. Sci. Rep. 2019, 9, 2514. [Google Scholar] [CrossRef]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jørgensen, H.; et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 2021, 118, 1713–1727. [Google Scholar] [CrossRef]
- Shi, H.; Mao, X.; Zhong, Y.; Liu, Y.; Zhao, X.; Yu, K.; Zhu, R.; Wei, Y.; Zhu, J.; Sun, H.; et al. Digoxin reduces atherosclerosis in apolipoprotein E-deficient mice. Br. J. Pharmacol. 2016, 173, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, I.; Oguiza, A.; Recio, C.; Mallavia, B.; Madrigal-Matute, J.; Blanco, J.; Egido, J.; Martin-Ventura, J.L.; Gomez-Guerrero, C. Targeting HSP90 Ameliorates Nephropathy and Atherosclerosis Through Suppression of NF-κB and STAT Signaling Pathways in Diabetic Mice. Diabetes 2015, 64, 3600–3613. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Lee, S.G.; Lim, S.; Jung, M.; Kwon, S.P.; Hong, J.; Kang, M.; Sohn, H.S.; Go, S.; Moon, S.; et al. A Senolytic-Eluting Coronary Stent for the Prevention of In-Stent Restenosis. ACS Biomater. Sci. Eng. 2022, 8, 1921–1929. [Google Scholar] [CrossRef]
- Nath, K.A.; O’Brien, D.R.; Croatt, A.J.; Grande, J.P.; Ackerman, A.W.; Nath, M.C.; Yamada, S.; Terzic, A.; Tchkonia, T.; Kirkland, J.L.; et al. The murine dialysis fistula model exhibits a senescence phenotype: Pathobiological mechanisms and therapeutic potential. Am. J. Physiol. Renal Physiol. 2018, 315, F1493–F1499. [Google Scholar] [CrossRef]
- Parvizi, M.; Franchi, F.; Arendt, B.K.; Ebtehaj, S.; Rodriguez-Porcel, M.; Lanza, I.R. Senolytic agents lessen the severity of abdominal aortic aneurysm in aged mice. Exp. Gerontol. 2021, 151, 111416. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021, 33, 818–832.e817. [Google Scholar] [CrossRef]
- Tateno, K.; Minamino, T.; Moriya, J.; Katada, A.; Yokoyama, M.; Miura, K.; Komuro, I. Cell therapy for cardiovascular diseases. Ann. Vasc. Dis. 2008, 1, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Toba, K.; Ozawa, T.; Kato, K.; Yanagawa, T.; Ikarashi, N.; Takayama, T.; Suzuki, T.; Hanawa, H.; Fuse, I.; et al. Establishment of culturing system for ex-vivo expansion of angiogenic immature erythroid cells, and its application for treatment of patients with chronic severe lower limb ischemia. J. Mol. Cell. Cardiol. 2010, 49, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Tateno, K.; Minamino, T.; Toko, H.; Akazawa, H.; Shimizu, N.; Takeda, S.; Kunieda, T.; Miyauchi, H.; Oyama, T.; Matsuura, K.; et al. Critical roles of muscle-secreted angiogenic factors in therapeutic neovascularization. Circ. Res. 2006, 98, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, D.E.; Forman, D.E. Cardiovascular care of older adults. Bmj 2021, 374, n1593. [Google Scholar] [CrossRef]
- Young, J.H.; Klag, M.J.; Muntner, P.; Whyte, J.L.; Pahor, M.; Coresh, J. Blood pressure and decline in kidney function: Findings from the Systolic Hypertension in the Elderly Program (SHEP). J. Am. Soc. Nephrol. 2002, 13, 2776–2782. [Google Scholar] [CrossRef]
- Izzo, J.L., Jr.; Levy, D.; Black, H.R. Clinical Advisory Statement. Importance of systolic blood pressure in older Americans. Hypertension 2000, 35, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Kihara, Y.; Noma, K. Endothelial dysfunction and hypertension in aging. Hypertens. Res. 2012, 35, 1039–1047. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Komoda, K.; Mikawa, R.; Asai, A.; Sugimoto, M. Cellular senescence promotes cancer metastasis by enhancing soluble E-cadherin production. iScience 2021, 24, 103022. [Google Scholar] [CrossRef]
- Asaithamby, A.; Shay, J.W.; Minna, J.D. Cellular senescence and lung cancer prognosis. Transl. Lung Cancer Res. 2022, 11, 1982–1987. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef]
- Mikawa, R.; Suzuki, Y.; Baskoro, H.; Kanayama, K.; Sugimoto, K.; Sato, T.; Sugimoto, M. Elimination of p19(ARF) -expressing cells protects against pulmonary emphysema in mice. Aging Cell 2018, 17, e12827. [Google Scholar] [CrossRef]
- Hashimoto, M.; Asai, A.; Kawagishi, H.; Mikawa, R.; Iwashita, Y.; Kanayama, K.; Sugimoto, K.; Sato, T.; Maruyama, M.; Sugimoto, M. Elimination of p19(ARF)-expressing cells enhances pulmonary function in mice. JCI Insight 2016, 1, e87732. [Google Scholar] [CrossRef]
- Suvakov, S.; Cubro, H.; White, W.M.; Butler Tobah, Y.S.; Weissgerber, T.L.; Jordan, K.L.; Zhu, X.Y.; Woollard, J.R.; Chebib, F.T.; Milic, N.M.; et al. Targeting senescence improves angiogenic potential of adipose-derived mesenchymal stem cells in patients with preeclampsia. Biol. Sex. Differ. 2019, 10, 49. [Google Scholar] [CrossRef]
- Suvakov, S.; Ghamrawi, R.; Cubro, H.; Tu, H.; White, W.M.; Tobah, Y.S.B.; Milic, N.M.; Grande, J.P.; Cunningham, J.M.; Chebib, F.T.; et al. Epigenetic and senescence markers indicate an accelerated ageing-like state in women with preeclamptic pregnancies. EBioMedicine 2021, 70, 103536. [Google Scholar] [CrossRef]
- Wyles, S.P.; Dashti, P.; Pirtskhalava, T.; Tekin, B.; Inman, C.; Gomez, L.S.; Lagnado, A.B.; Prata, L.; Jurk, D.; Passos, J.F.; et al. A chronic wound model to investigate skin cellular senescence. Aging 2023, 15, 2852–2862. [Google Scholar] [CrossRef]
- Dańczak-Pazdrowska, A.; Gornowicz-Porowska, J.; Polańska, A.; Krajka-Kuźniak, V.; Stawny, M.; Gostyńska, A.; Rubiś, B.; Nourredine, S.; Ashiqueali, S.; Schneider, A.; et al. Cellular senescence in skin-related research: Targeted signaling pathways and naturally occurring therapeutic agents. Aging Cell 2023, e13845. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L. Tumor dormancy and disease recurrence. Cancer Metastasis Rev. 2023, 42, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Olson, I.; Mansour, M.; Carlstrom, L.P.; Sutiwisesak, R.; Saber, R.; Rajani, K.; Warrington, A.E.; Howard, A.; Schroeder, M.; et al. Selective Vulnerability of Senescent Glioblastoma Cells to BCL-XL Inhibition. Mol. Cancer Res. 2022, 20, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef] [PubMed]


| Cell Type | SASP Factors | Possible Cell-Type Specific Senescence Markers and Hallmarks |
|---|---|---|
| Endothelial cells | IL-1, IL-6, IL-8, Il-15, MCP-1, TNF-α, SELP, ANGPTL-2, HTRA4, PCNA, CHEK4, and VEGF | Down-regulation of nitric oxide (NO) and eNOS |
| Prothrombotic metalloprotease (PAI-1, and PAI-2) activity | ||
| Expression of adhesion molecules (ICAM-1, VCAM, and PECAM-1) | ||
| Production of Angiotensin II (Ang II) and Endothelin 1 | ||
| IGF binding protein expression, such as IGFBP-5 | ||
| microRNA34a | ||
| Vascular smooth muscle cells | IL-1a, IL-6, IL-8, TGF-ß1, and MMPs, CCL-3/CCL-4, and MCP-1 | Calcification markers (OPN, OPG, Runx-2, BMPs, and ALP) ↑ |
| Fibrosis markers (elastase ↑, collagen ↓) | ||
| Muscle contraction markers (α-SMA ↓, SM-MHC ↓and calponin ↓) | ||
| microRNA34a | ||
| Cardiomyocytes | IL-6, EDN3, TGF-ß2 and GDF15 | Fatty acid oxidation enzyme (CPT1) activity ↓ |
| Glutathione reductase activity ↓ | ||
| PPARα and PGC-1α activity ↓ | ||
| Troponin I phosphorylation ↓ | ||
| Macrophages | IL-1ß, IL-6, TNF-α, and MMP-3, and MMP-13 | TM1, SIPR1, TAM, MERTK and BRD4 |
| T cells | IL-6, TNF-α, IFN- γ and OPN | Perforins and granzymes ↑ |
| Down-regulation of CD27 and CD28 | ||
| CD4+ CD44high CD62Llow PD-1+ CD153+ | ||
| Fibroblasts | Myofibroblast markers (POSTN, PARP1, IL-6, IL-13, THBS1, and TGF-ß1) | |
| Matricellular protein (CCN1) ↑ |
| Cell Type | Initiators | Pathology | Diseases |
| Endothelial cells | Telomere shortening Shear stress from blood flow Dyslipidemia AngII signaling LDL oxidation Hyperinsulinemia Doxorubicin Irradiation ROS | Local and systemic inflammation Impaired vascular relaxation Hindered blood flow regulation Hypercoagulability Angiogenesis ↓ Expression of adhesion molecules ↑ | Hypertension Atherosclerosis Peripheral artery disease Ischemic heart disease |
| Vascular smooth muscle cells | Telomeric dysfunction AngII signaling Dyslipidemia Hyperinsulinemia ROS | Vascular calcification Fibrosis Impaired smooth muscle contraction Hindered blood flow regulation Atherosclerotic plaque vulnerability | Hypertension Aortic aneurysm Aortic dissection Atherosclerosis Myocardial infarction |
| Cardiomyocytes | Pressure overload Hypoxia Cardiotoxic drugs Irradiation ROS | Cardiac dysfunction Cardiac hypertrophy Fibrosis | Heart failure Cardiomyopathy Ischemic heart disease Arrhythmias |
| Immune cells | Dyslipidemia Cell debris ROS | Inflammation Impaired cardiac electrical conduction | Atherosclerosis Heart failure Arrhythmias |
| Fibroblasts | Pressure overload Hypoxia Cardiotoxic drugs Irradiation ROS | Fibrosis Cardiac dysfunction Cardiac hypertrophy | Heart failure Cardiomyopathy Arrhythmias |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suda, M.; Paul, K.H.; Minamino, T.; Miller, J.D.; Lerman, A.; Ellison-Hughes, G.M.; Tchkonia, T.; Kirkland, J.L. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells 2023, 12, 1296. https://doi.org/10.3390/cells12091296
Suda M, Paul KH, Minamino T, Miller JD, Lerman A, Ellison-Hughes GM, Tchkonia T, Kirkland JL. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells. 2023; 12(9):1296. https://doi.org/10.3390/cells12091296
Chicago/Turabian StyleSuda, Masayoshi, Karl H. Paul, Tohru Minamino, Jordan D. Miller, Amir Lerman, Georgina M. Ellison-Hughes, Tamar Tchkonia, and James L. Kirkland. 2023. "Senescent Cells: A Therapeutic Target in Cardiovascular Diseases" Cells 12, no. 9: 1296. https://doi.org/10.3390/cells12091296