
IKKβ Inhibition Attenuates Epithelial Mesenchymal Transition of Human Stem Cell-Derived Retinal Pigment Epithelium
 , , , , and
, , , , and 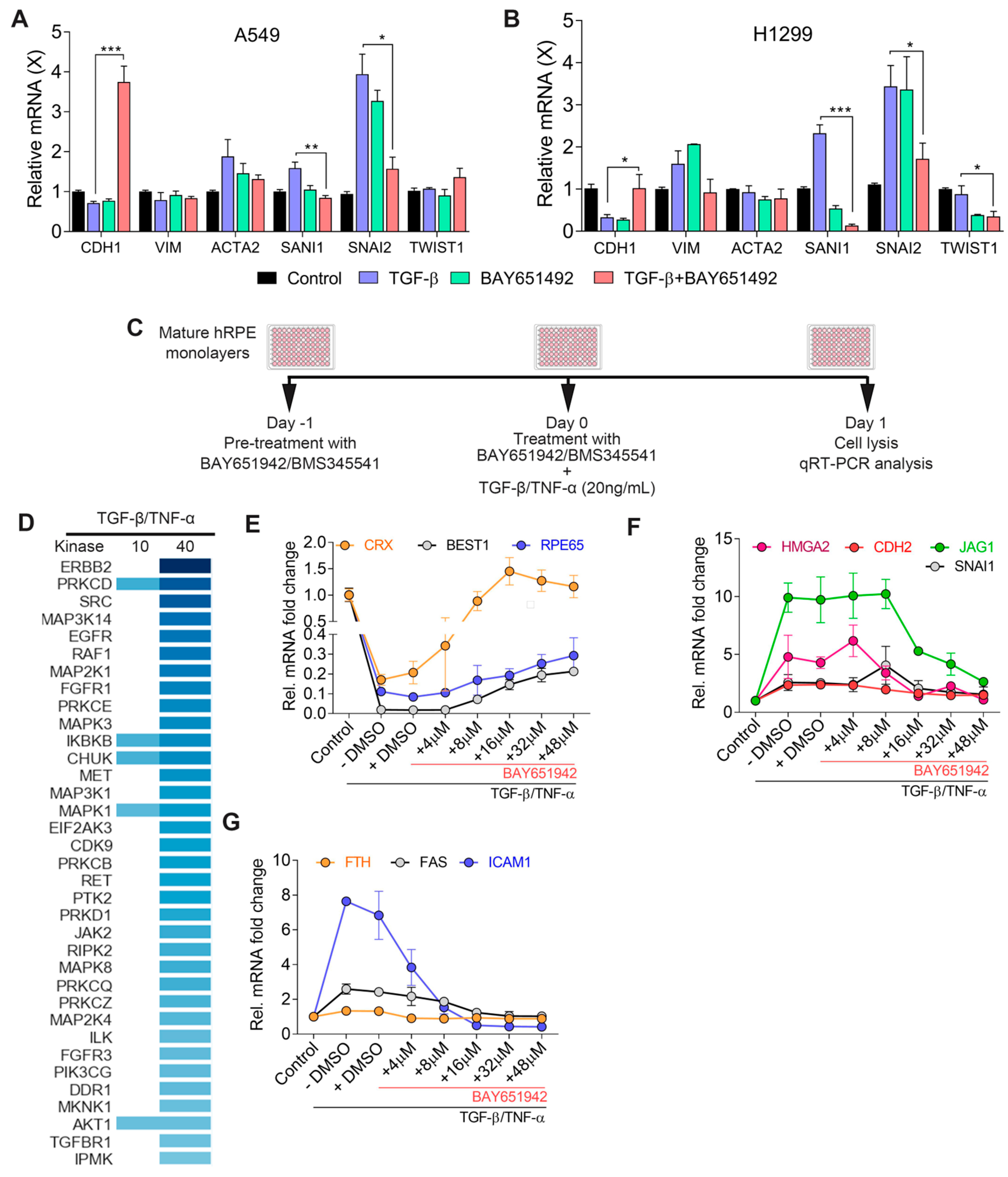{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Pluripotent/Embryonic Stem Cell (hiPSC/hESC) Cultures and RPE Differentiation
2.2. A549 and H1299 Cell Cultures and EMT Induction In Vitro
2.3. Small-Molecule Treatment
2.4. RPE–EMT Induction In Vitro
2.5. RNA Isolation and Quantitative RT-PCR (qRT-PCR)
2.6. RNA-Seq and Data Processing
2.7. RNA-Seq Data Analysis
2.8. Biological Pathway and Upstream Regulator Analysis
2.9. Hierarchical Transcription Factors Analysis
2.10. Statistical Analysis
3. Results
3.1. IKKβ Inhibitor (BAY651942) Treatment Partially Inhibits TGF–β/TNF–α-Induced RPE–EMT and Restores Expression of RPE Genes
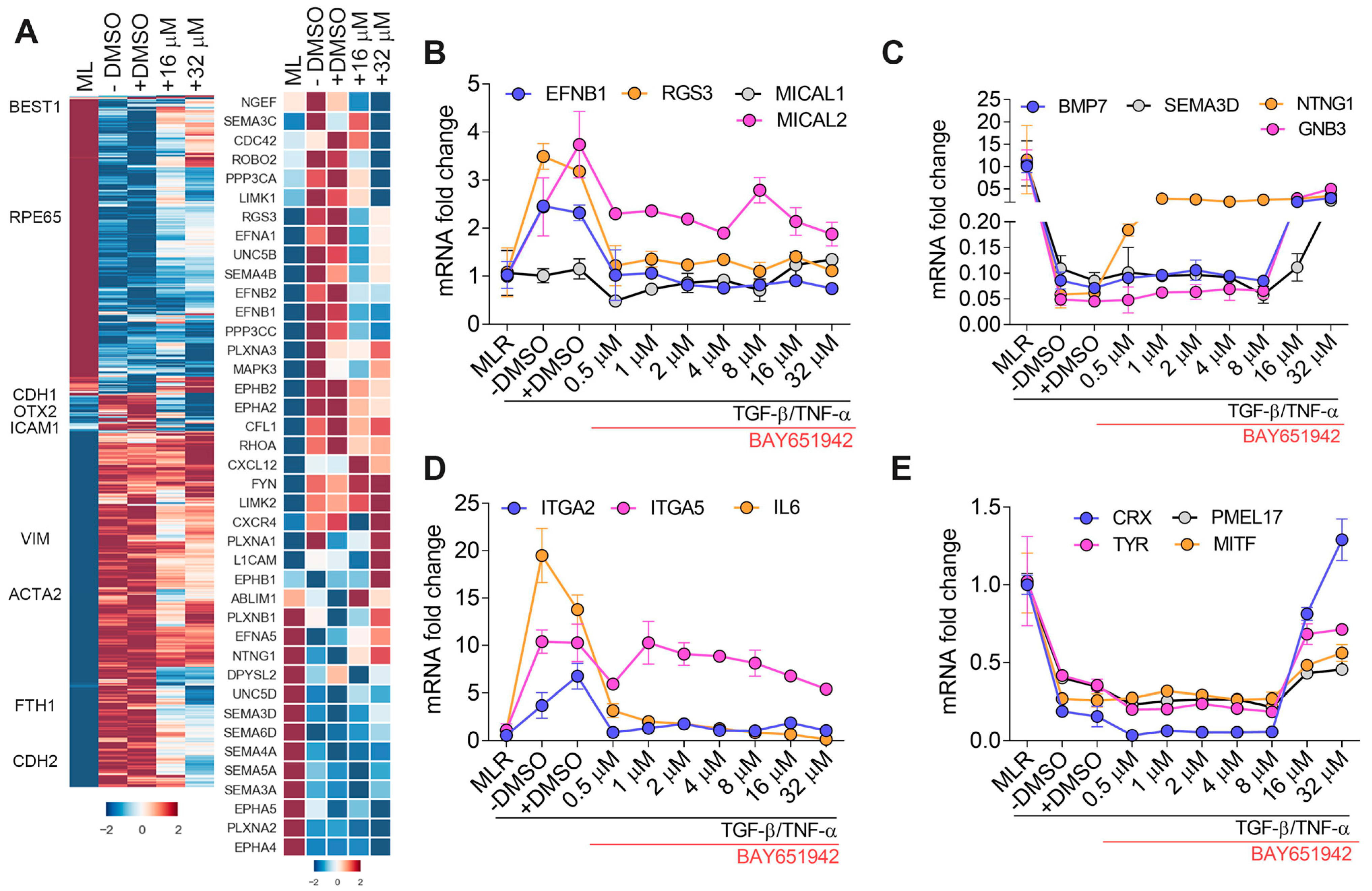
3.2. BAY651942 Restores Normalized Expression of Multiple Axon Guidance Molecules
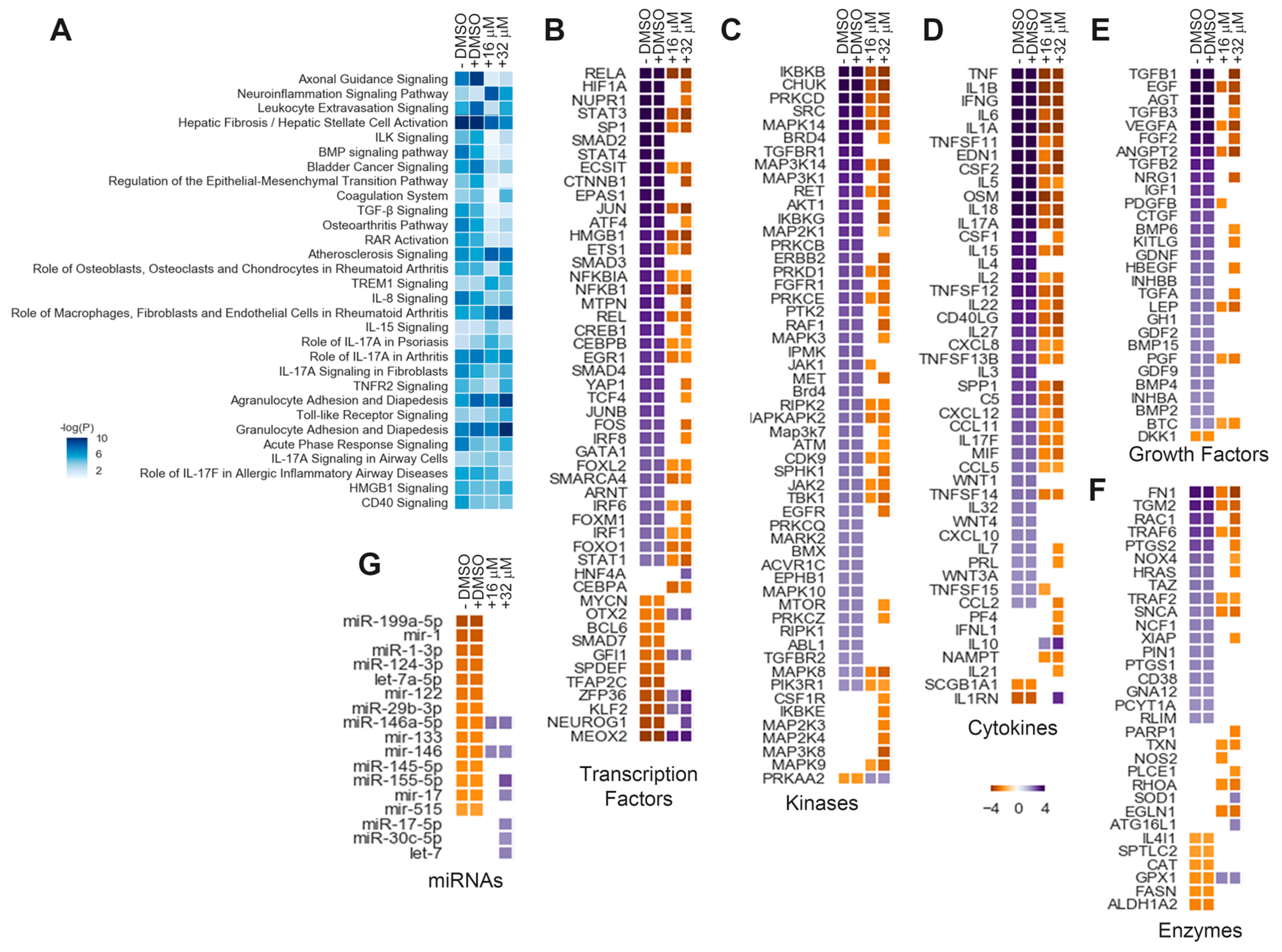
3.3. BAY651942 Modulates Multiple RPE–EMT-Associated Biological Pathways and Upstream Regulators
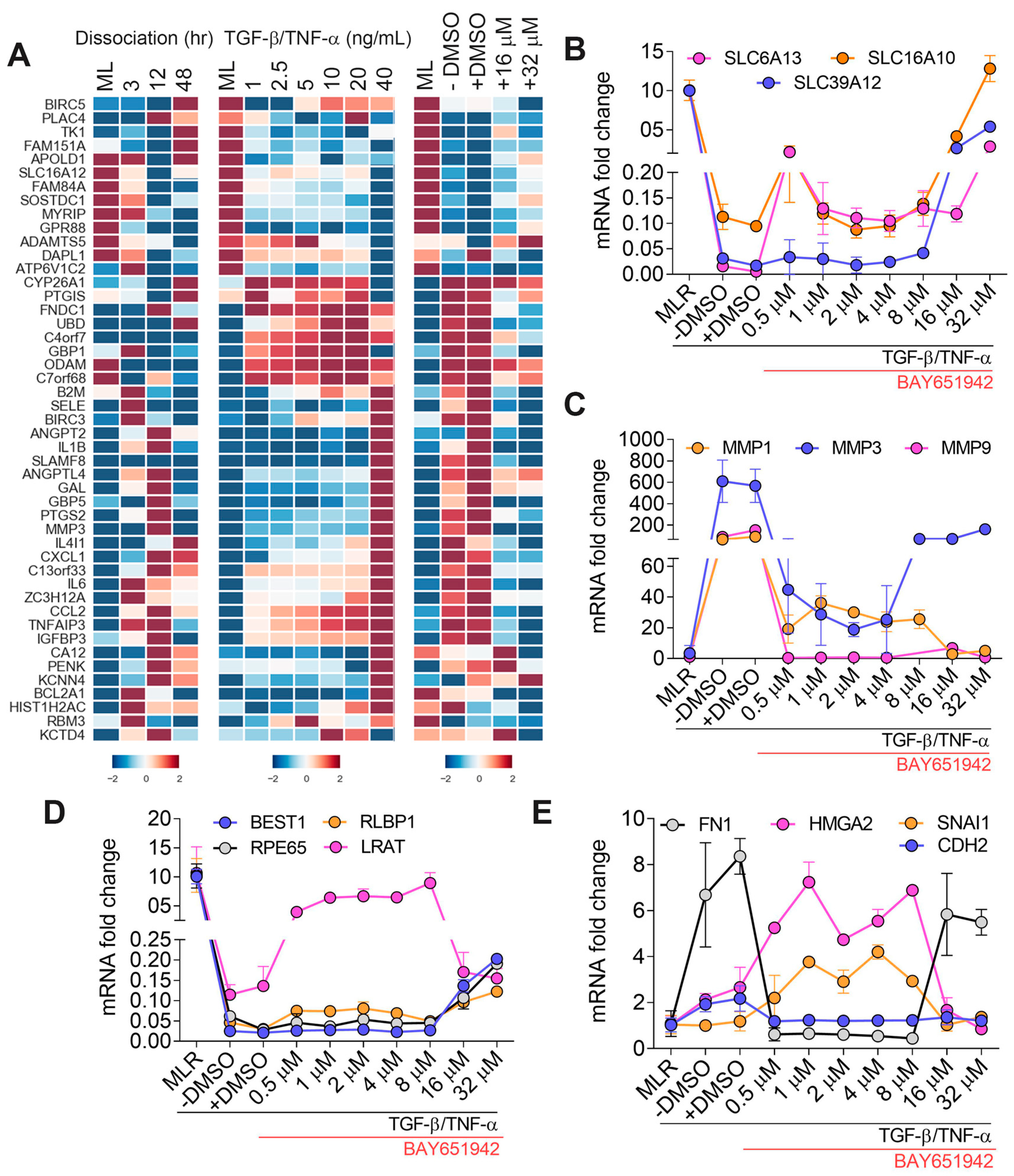
3.4. BAY651942 Restores Multiple AMD-Associated Risk Factors That Were Altered by TGF–β/TNF–α-Induced RPE–EMT
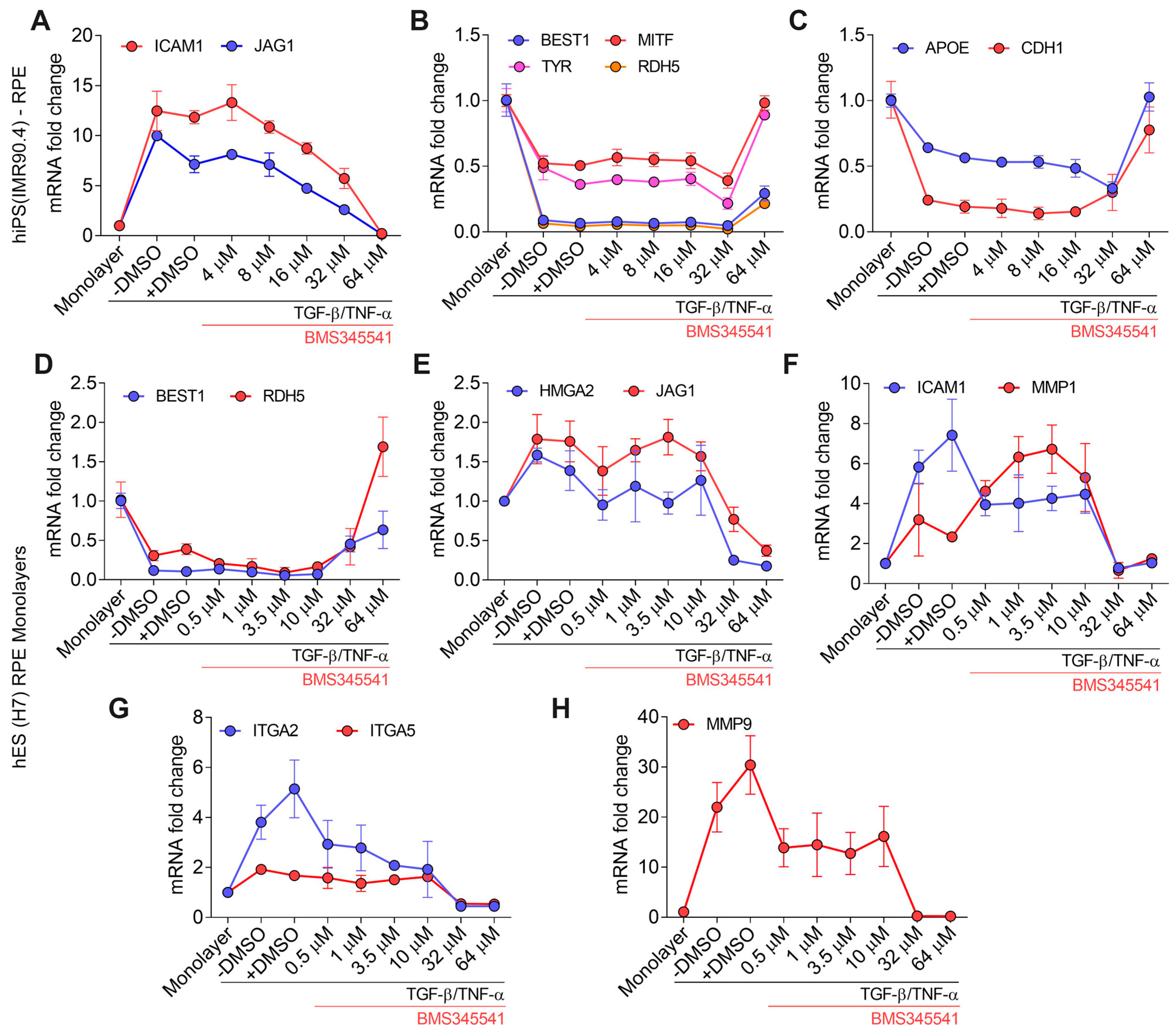
3.5. Validation of TGF–β/TNF–α-Induced RPE–EMT Using IKKβ Inhibitor (BMS345541)
3.6. Hierarchial Analysis of Transcription Factors Affected by Inhibition of NFκB Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhou, J.; Li, D. Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 2021, 12, 727870. [Google Scholar] [CrossRef]
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelium Dysfunction. Front. Cell Dev. Biol. 2020, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef]
- Tamiya, S.; Kaplan, H.J. Role of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Exp. Eye Res. 2016, 142, 26–31. [Google Scholar] [CrossRef]
- Boles, N.C.; Fernandes, M.; Swigut, T.; Srinivasan, R.; Schiff, L.; Rada-Iglesias, A.; Wang, Q.; Saini, J.S.; Kiehl, T.; Stern, J.H.; et al. Epigenomic and Transcriptomic Changes During Human RPE EMT in a Stem Cell Model of Epiretinal Membrane Pathogenesis and Prevention by Nicotinamide. Stem Cell Rep. 2020, 14, 631–647. [Google Scholar] [CrossRef]
- Ghosh, S.; Shang, P.; Terasaki, H.; Stepicheva, N.; Hose, S.; Yazdankhah, M.; Weiss, J.; Sakamoto, T.; Bhutto, I.A.; Xia, S.; et al. A Role for betaA3/A1-Crystallin in Type 2 EMT of RPE Cells Occurring in Dry Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD104–AMD113. [Google Scholar] [CrossRef] [Green Version]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Sripathi, S.R.; Hu, M.W.; Turaga, R.C.; Mertz, J.; Liu, M.M.; Wan, J.; Maruotti, J.; Wahlin, K.J.; Berlinicke, C.A.; Qian, J.; et al. Proteome Landscape of Epithelial-to-Mesenchymal Transition (EMT) of Retinal Pigment Epithelium Shares Commonalities With Malignancy-Associated EMT. Mol. Cell Proteom. 2021, 20, 100131. [Google Scholar] [CrossRef] [PubMed]
- Sripathi, S.R.; Hu, M.W.; Liu, M.M.; Wan, J.; Cheng, J.; Duan, Y.; Mertz, J.L.; Wahlin, K.J.; Maruotti, J.; Berlinicke, C.A.; et al. Transcriptome Landscape of Epithelial to Mesenchymal Transition of Human Stem Cell-Derived RPE. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.L.; Sripathi, S.R.; Yang, X.; Chen, L.; Esumi, N.; Zhang, H.; Zack, D.J. Proteomic and phosphoproteomic analyses identify liver-related signaling in retinal pigment epithelial cells during EMT. Cell Rep. 2021, 37, 109866. [Google Scholar] [CrossRef] [PubMed]
- Casaroli-Marano, R.P.; Pagan, R.; Vilaro, S. Epithelial-mesenchymal transition in proliferative vitreoretinopathy: Intermediate filament protein expression in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2062–2072. [Google Scholar]
- Hirasawa, M.; Noda, K.; Noda, S.; Suzuki, M.; Ozawa, Y.; Shinoda, K.; Inoue, M.; Ogawa, Y.; Tsubota, K.; Ishida, S. Transcriptional factors associated with epithelial-mesenchymal transition in choroidal neovascularization. Mol. Vis. 2011, 17, 1222–1230. [Google Scholar]
- Radeke, M.J.; Radeke, C.M.; Shih, Y.H.; Hu, J.; Bok, D.; Johnson, L.V.; Coffey, P.J. Restoration of mesenchymal retinal pigmented epithelial cells by TGFbeta pathway inhibitors: Implications for age-related macular degeneration. Genome Med. 2015, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Leong, B.; Messinger, J.D.; Kar, D.; Ach, T.; Yannuzzi, L.A.; Freund, K.B.; Curcio, C.A. Hyperreflective Foci, Optical Coherence Tomography Progression Indicators in Age-Related Macular Degeneration, Include Transdifferentiated Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2021, 62, 34. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat. Rev. Immunol. 2002, 2, 664–674. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; George, A.; Nimmagadda, M.; Ortolan, D.; Karla, B.S.; Qureshy, Z.; Bose, D.; Dejene, R.; Liang, G.; Wan, Q.; et al. Epithelial phenotype restoring drugs suppress macular degeneration phenotypes in an iPSC model. Nat. Commun. 2021, 12, 7293. [Google Scholar] [CrossRef]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005, 145, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Qian, L.; Flood, P.M.; Shi, J.S.; Hong, J.S.; Gao, H.M. Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2010, 333, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhise, N.S.; Wahlin, K.J.; Zack, D.J.; Green, J.J. Evaluating the potential of poly(beta-amino ester) nanoparticles for reprogramming human fibroblasts to become induced pluripotent stem cells. Int. J. Nanomed. 2013, 8, 4641–4658. [Google Scholar]
- Maruotti, J.; Wahlin, K.; Gorrell, D.; Bhutto, I.; Lutty, G.; Zack, D.J. A simple and scalable process for the differentiation of retinal pigment epithelium from human pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 341–354. [Google Scholar] [CrossRef]
- Maruotti, J.; Sripathi, S.R.; Bharti, K.; Fuller, J.; Wahlin, K.J.; Ranganathan, V.; Sluch, V.M.; Berlinicke, C.A.; Davis, J.; Kim, C.; et al. Small-molecule-directed, efficient generation of retinal pigment epithelium from human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10950–10955. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Wang, J.; Zibetti, C.; Shang, P.; Sripathi, S.R.; Zhang, P.; Cano, M.; Hoang, T.; Xia, S.; Ji, H.; Merbs, S.L.; et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 2018, 9, 1364. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Lin, J.; Zack, D.J.; Mendell, J.T.; Qian, J. Analysis of regulatory network topology reveals functionally distinct classes of microRNAs. Nucleic Acids Res. 2008, 36, 6494–6503. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lin, J.; Masuda, T.; Esumi, N.; Zack, D.J.; Qian, J. Genome-wide prediction and characterization of interactions between transcription factors in Saccharomyces cerevisiae. Nucleic Acids Res. 2006, 34, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Kuriyan, A.E.; Su, C.W.; Mahabole, M.; Zhang, Y.; Zhu, Y.T.; Flynn, H.W.; Parel, J.M.; Tseng, S.C. Inhibition of Proliferation and Epithelial Mesenchymal Transition in Retinal Pigment Epithelial Cells by Heavy Chain-Hyaluronan/Pentraxin 3. Sci. Rep. 2017, 7, 43736. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, D.; Yang, S.; Yao, H.; Li, M.; Zhao, C.; Zhang, J.; Xu, G.T.; Li, H.; Wang, F. Protective Effects of Fucoidan on Epithelial-Mesenchymal Transition of Retinal Pigment Epithelial Cells and Progression of Proliferative Vitreoretinopathy. Cell. Physiol. Biochem. 2018, 46, 1704–1715. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Tokuda, K.; Yamashiro, C.; Higashijima, F.; Yoshimoto, T.; Ota, M.; Ogata, T.; Ashimori, A.; Hatano, M.; Kobayashi, M.; et al. Inhibition of epithelial-mesenchymal transition in retinal pigment epithelial cells by a retinoic acid receptor-alpha agonist. Sci. Rep. 2021, 11, 11842. [Google Scholar] [CrossRef]
- Ishikawa, K.; He, S.; Terasaki, H.; Nazari, H.; Zhang, H.; Spee, C.; Kannan, R.; Hinton, D.R. Resveratrol inhibits epithelial-mesenchymal transition of retinal pigment epithelium and development of proliferative vitreoretinopathy. Sci. Rep. 2015, 5, 16386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lapidus, R.G.; Liu, P.; Choi, E.Y.; Adediran, S.; Hussain, A.; Wang, X.; Liu, X.; Dan, H.C. Targeting IkappaB Kinase beta/NF-kappaB Signaling in Human Prostate Cancer by a Novel IkappaB Kinase beta Inhibitor CmpdA. Mol. Cancer Ther. 2016, 15, 1504–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, H.; Yang, F.; Wang, M.; Niu, Y.; Xing, N. IKK inhibitor suppresses epithelial-mesenchymal transition and induces cell death in prostate cancer. Oncol. Rep. 2016, 36, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Maier, H.J.; Alacakaptan, M.; Wiedemann, E.; Braunger, J.; Boehmelt, G.; Madwed, J.B.; Young, E.R.; Marshall, D.R.; Pehamberger, H.; et al. BI 5700, a Selective Chemical Inhibitor of IkappaB Kinase 2, Specifically Suppresses Epithelial-Mesenchymal Transition and Metastasis in Mouse Models of Tumor Progression. Genes Cancer 2010, 1, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Yang, Z.; Carter-Cooper, B.; Chang, E.T.; Choi, E.Y.; Kallakury, B.; Liu, X.; Lapidus, R.G.; Cullen, K.J.; Dan, H. Suppression of migration, invasion, and metastasis of cisplatin-resistant head and neck squamous cell carcinoma through IKKbeta inhibition. Clin. Exp. Metastasis 2020, 37, 283–292. [Google Scholar] [CrossRef]
- Tsolou, A.; Liousia, M.; Kalamida, D.; Pouliliou, S.; Giatromanolaki, A.; Koukourakis, M. Inhibition of IKK-NFkappaB pathway sensitizes lung cancer cell lines to radiation. Cancer Biol. Med. 2017, 14, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Kyakulaga, A.H.; Aqil, F.; Munagala, R.; Gupta, R.C. Withaferin A inhibits Epithelial to Mesenchymal Transition in Non-Small Cell Lung Cancer Cells. Sci. Rep. 2018, 8, 15737. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.I.; Debski, K.J.; Dabrowski, M.; Czarnecka, A.M.; Szczylik, C. Gene set enrichment analysis and ingenuity pathway analysis of metastatic clear cell renal cell carcinoma cell line. Am. J. Physiol. Ren. Physiol. 2016, 311, F424–F436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Gallo, N.B.; Hancox, L.S.; Miller, N.J.; Radeke, C.M.; Maloney, M.A.; Cooper, J.B.; Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med. 2012, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Daniele, L.L.; Sauer, B.; Gallagher, S.M.; Pugh, E.N., Jr.; Philp, N.J. Altered visual function in monocarboxylate transporter 3 (Slc16a8) knockout mice. Am. J. Physiol. Cell Physiol. 2008, 295, C451–C457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, N.G.; ElShelmani, H.; Singh, M.K.; Mansergh, F.C.; Wride, M.A.; Padilla, M.; Keegan, D.; Hogg, R.E.; Ambati, B.K. Risk factors and biomarkers of age-related macular degeneration. Prog. Retin. Eye Res. 2016, 54, 64–102. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Oliver, E.; Maratou, K.; Atanur, S.S.; Dubois, O.D.; Cotroneo, E.; Chen, C.N.; Wang, L.; Arce, C.; Chabosseau, P.L.; et al. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature 2015, 524, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Booij, J.C.; ten Brink, J.B.; Swagemakers, S.M.; Verkerk, A.J.; Essing, A.H.; van der Spek, P.J.; Bergen, A.A. A new strategy to identify and annotate human RPE-specific gene expression. PLoS ONE 2010, 5, e9341. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, S.S.; Braun, T.A.; Skeie, J.M.; Haas, C.M.; Sohn, E.H.; Stone, E.M.; Scheetz, T.E.; Mullins, R.F. Altered gene expression in dry age-related macular degeneration suggests early loss of choroidal endothelial cells. Mol. Vis. 2013, 19, 2274–2297. [Google Scholar]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, C.H.; Occleston, N.L.; Charteris, D.; Daniels, J.; Aylward, G.W.; Khaw, P.T. A prospective study of matrix metalloproteinases in proliferative vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1524–1529. [Google Scholar]
- Nita, M.; Strzalka-Mrozik, B.; Grzybowski, A.; Mazurek, U.; Romaniuk, W. Age-related macular degeneration and changes in the extracellular matrix. Med. Sci. Monit. 2014, 20, 1003–1016. [Google Scholar] [PubMed] [Green Version]
- Liutkeviciene, R.; Lesauskaite, V.; Sinkunaite-Marsalkiene, G.; Zaliuniene, D.; Zaliaduonyte-Peksiene, D.; Mizariene, V.; Gustiene, O.; Jasinskas, V.; Jariene, G.; Tamosiunas, A. The Role of Matrix Metalloproteinases Polymorphisms in Age-Related Macular Degeneration. Ophthalmic Genet. 2015, 36, 149–155. [Google Scholar] [CrossRef]
- Lin, C.Y.; Tsai, P.H.; Kandaswami, C.C.; Lee, P.P.; Huang, C.J.; Hwang, J.J.; Lee, M.T. Matrix metalloproteinase-9 cooperates with transcription factor Snail to induce epithelial-mesenchymal transition. Cancer Sci. 2011, 102, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Kawa, M.P.; Machalinska, A.; Roginska, D.; Machalinski, B. Complement system in pathogenesis of AMD: Dual player in degeneration and protection of retinal tissue. J. Immunol. Res. 2014, 2014, 483960. [Google Scholar] [CrossRef] [Green Version]
- Jadeja, R.N.; Powell, F.L.; Jones, M.A.; Fuller, J.; Joseph, E.; Thounaojam, M.C.; Bartoli, M.; Martin, P.M. Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging 2018, 10, 1306–1323. [Google Scholar] [CrossRef]
- Su, P.; Hu, J.; Zhang, H.; Jia, M.; Li, W.; Jing, X.; Zhou, G. Association of TRPS1 gene with different EMT markers in ERalpha-positive and ERalpha-negative breast cancer. Diagn. Pathol. 2014, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Sripathi, S.R.; Liu, M.M.; Hu, M.-W.; Wan, J.; Cheng, J.; Duan, Y.; Mertz, J.; Wahlin, K.; Maruotti, J.; Berlinicke, C.A.; et al. Axon Guidance Signaling Modulates Epithelial to Mesenchymal Transition in Stem Cell-Derived Retinal Pigment Epithelium. bioRxiv 2018, 264705. [Google Scholar] [CrossRef]
- Yang, X.; Rai, U.; Chung, J.Y.; Esumi, N. Fine Tuning of an Oxidative Stress Model with Sodium Iodate Revealed Protective Effect of NF-kappaB Inhibition and Sex-Specific Difference in Susceptibility of the Retinal Pigment Epithelium. Antioxidants 2021, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Wang, S. Not All Stressors Are Equal: Mechanism of Stressors on RPE Cell Degeneration. Front. Cell Dev. Biol. 2020, 8, 591067. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kang, K.H.; Burrola, P.; Mak, T.W.; Lemke, G. Retinal degeneration triggered by inactivation of PTEN in the retinal pigment epithelium. Genes Dev. 2008, 22, 3147–3157. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chung, J.Y.; Rai, U.; Esumi, N. Cadherins in the retinal pigment epithelium (RPE) revisited: P-cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PLoS ONE 2018, 13, e0191279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef] [Green Version]
- McLeod, D.S.; Grebe, R.; Bhutto, I.; Merges, C.; Baba, T.; Lutty, G.A. Relationship between RPE and choriocapillaris in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4982–4991. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Lu, Q.; Gaddipati, S.; Kasetti, R.B.; Wang, W.; Pasparakis, M.; Kaplan, H.J.; Li, Q. IKK2 inhibition attenuates laser-induced choroidal neovascularization. PLoS ONE 2014, 9, e87530. [Google Scholar] [CrossRef]
- Moss, N.C.; Stansfield, W.E.; Willis, M.S.; Tang, R.H.; Selzman, C.H. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2248–H2253. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xin, Y.; Ye, F.; Wang, W.; Lu, Q.; Kaplan, H.J.; Dean, D.C. Taz-tead1 links cell-cell contact to zeb1 expression, proliferation, and dedifferentiation in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3372–3378. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, S.; Zou, C.; Levine, E.M. Retinal pigment epithelium development, plasticity, and tissue homeostasis. Exp. Eye Res. 2014, 123, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Bharti, K.; Nguyen, M.T.; Skuntz, S.; Bertuzzi, S.; Arnheiter, H. The other pigment cell: Specification and development of the pigmented epithelium of the vertebrate eye. Pigment Cell Res. 2006, 19, 380–394. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sripathi, S.R.; Hu, M.-W.; Turaga, R.C.; Mikeasky, R.; Satyanarayana, G.; Cheng, J.; Duan, Y.; Maruotti, J.; Wahlin, K.J.; Berlinicke, C.A.; et al. IKKβ Inhibition Attenuates Epithelial Mesenchymal Transition of Human Stem Cell-Derived Retinal Pigment Epithelium. Cells 2023, 12, 1155. https://doi.org/10.3390/cells12081155
Sripathi SR, Hu M-W, Turaga RC, Mikeasky R, Satyanarayana G, Cheng J, Duan Y, Maruotti J, Wahlin KJ, Berlinicke CA, et al. IKKβ Inhibition Attenuates Epithelial Mesenchymal Transition of Human Stem Cell-Derived Retinal Pigment Epithelium. Cells. 2023; 12(8):1155. https://doi.org/10.3390/cells12081155
Chicago/Turabian StyleSripathi, Srinivasa R., Ming-Wen Hu, Ravi Chakra Turaga, Rebekah Mikeasky, Ganesh Satyanarayana, Jie Cheng, Yukan Duan, Julien Maruotti, Karl J. Wahlin, Cynthia A. Berlinicke, and et al. 2023. "IKKβ Inhibition Attenuates Epithelial Mesenchymal Transition of Human Stem Cell-Derived Retinal Pigment Epithelium" Cells 12, no. 8: 1155. https://doi.org/10.3390/cells12081155