1. Introduction
MicroRNAs (miRNAs) are a class of single-stranded noncoding RNA molecules containing 17–24 nucleotides [
1,
2,
3]. The first miRNA, lin-4, and the first mammalian miRNA, let-7, were found in the 1990s [
4,
5]. With these two significant discoveries, a wave of genomic research took place, resulting in the discovery of a large number of miRNAs in many organisms [
6,
7]. At the same time, it has become increasingly evident to researchers that miRNAs are involved in complex and diverse life processes. Specifically, miRNAs can bind to complementary target mRNAs, resulting in mRNA translational inhibition or degradation, which means that miRNAs have a significant impact on cell differentiation, proliferation, and apoptosis [
1]. In addition, miRNAs play essential roles in various cellular activities, including immune responses and neurotransmitter synthesis [
8,
9]. More significantly, miRNAs participate in tumorigenesis and host–pathogen interactions [
10,
11,
12,
13]. For instance, Liu et al. [
14] identified that the abnormal expression of miR-181c is involved in the pathogenesis of glioblastoma. Therefore, restoring the expression level of miR-181c in glioblastoma cancer cells can effectively treat the disease, which also provides new insight for the clinical treatment of many refractory diseases, including cancer.
Indeed, as a low-molecular-weight compound, small molecule (SM) drugs have been demonstrated to target dysregulated miRNAs and modulate their expression [
15,
16,
17]. For instance, SPC3649, the first miRNA-targeted drug administered in human clinical trials, successfully inhibits the expression of miR-122, which is required for hepatitis C virus replication [
18]. Consequently, utilizing miRNAs as diagnostic and therapeutic targets has become a promising pathway in drug development and disease treatment. Since developing new SMs is time-consuming and expensive, it is extremely difficult to develop specific SMs for each dysregulated miRNA. Therefore, researchers can look into utilizing existing SMs to target and modulate a wider variety of miRNAs [
19]. Furthermore, determining the potential associations between the known SMs and miRNAs through biological experiments is of great significance and urgency.
Given the abundance of existing SMs and miRNAs, it is critical to pre-screen out SM–miRNA samples with high association probabilities for complex and expensive biological experiments. The proposed predictive approaches may be divided into network inference-based models and matrix-completion-based models. For the first kind of model, Guan et al. [
20] proposed a model called Graphlet Interaction-based Inference for Small Molecule–miRNA Association Prediction (GISMMA). Based on the integrated SM/miRNA similarity (the widely used SM/miRNA similarities include the side-effect-based SM similarity, the chemical-structure-based SM similarity, the functional consistency-based SM similarity, the indication phenotype-based SM similarity, the gene functional consistency-based miRNA similarity, and the disease-phenotype-based miRNA similarity), they first constructed the SM/miRNA similarity network. Then, a specific SM–miRNA association score was calculated by counting the number of graphlet interactions throughout the SM/miRNA similarity network. Furthermore, Li et al. [
21] developed the Small Molecule–miRNA Network-Based Inference (SMiR-NBI) predictive model. In a constructed SM–miRNA heterogeneous network, a given SM node evenly distributes the obtained initial resources to the miRNA nodes regulated by it. Following this, these miRNA nodes immediately distribute the obtained resources to the SM nodes adjacent to them. As the resources are continuously propagated through the network, the resource allocation of all nodes eventually stabilizes. The final resource fraction of the miRNA nodes reflects the possibility of being regulated by the given SM. Notably, the model is incapable of predicting miRNAs or SMs that are potentially associated with new SMs or miRNAs. Additionally, Qu et al. [
22] proposed the Triple Layer Heterogeneous Network-based Small Molecule–miRNA Association (TLHNSMMA) predictive model. They first constructed an SM–miRNA-disease triple layer heterogeneous network. An iterative update algorithm was then applied to obtain the association scores of all SM–miRNA pairs. Benefiting from the introduction of additional information, the model demonstrated excellent prediction accuracy. However, it is likewise not applicable to predict miRNAs or SMs that are potentially associated with new SMs or miRNAs. In view of the unreliability of all the aforementioned methods due to the presence of noise data, Yin et al. [
23] developed a new computational method called Sparse Learning and Heterogeneous Graph Inference for Small Molecule–miRNA Associations (SLHGISMMA) prediction. They first decomposed the SM–miRNA association matrix into two parts, in which the first part is a linear combination of the original association matrix and a low-rank matrix, and the second part is a sparse noise matrix. After eliminating the noise matrix, they integrated the SM/miRNA similarity information and the information in the reacquired association matrix into a heterogeneous graph. Finally, the association scores were obtained by implementing a heterogeneous graph inference algorithm. The drawback of SLHGISMMA is that it cannot restrict the prediction scores in [0, 1], which reduces the interpretability and accuracy of the association scores.
Additionally, several matrix-completion-based heuristic algorithms are likewise applied in predicting potential SM–miRNA associations. Inspired by the traditional CMF [
24] method, Wang et al. [
25] developed a model called Dual-Network Collaborative Matrix Factorization (DCMF) for predicting small molecule–miRNA associations. They first preprocessed the SM–miRNA association matrix utilizing the Weighted K-Nearest Known Neighbors (WKNKN) method. In addition to the Tikhonov regularization term, they incorporated two new regularization terms in the optimization framework of the traditional matrix factorization model. After solving the optimization problem, they calculated the matmul product of the two low-rank feature matrices as the completed matrix, and the completed values were considered as the association scores. Moreover, a model named Predicting Potential Small Molecule–miRNA Associations based on Bounded Nuclear Norm Regularization (BNNRSMMA) was developed by Chen et al. [
26]. They recovered the target matrix with the missing values by minimizing its nuclear norm. Although BNNRSMMA restricts the completed value in [0, 1], it may obtain a highly biased solution as the nuclear norm may not be the optimal convex approximation to the rank function, and the prediction accuracy cannot be guaranteed. Considering that the rank of the result matrix is non-adjustable, this decreases the adaptability of BNNRSMMA on different datasets. The main innovative points and limitations of the above models are shown in
Table 1.
Considering that the previous models have some limitations, we develop a more accurate predictive model called AMCSMMA, which overcomes the insufficiencies listed in
Table 1, and its framework is shown in
Figure 1. In Validation Experiment A, the AUC scores achieve the best results with values ranging from 0.9974 to 0.9981 when parameter
, and we finally set
to reduce the computational complexity. Additionally, we design Validation Experiment B in which the values of the AUC, Precision, Recall, F1 Score, Accuracy, and MCC are all above 0.97. Moreover, we conduct four types of cross-validation (CV) experiments based on Dataset 1 (Dataset 2). As a result, the AUC values of AMCSMMA are 0.9910 ± 0.0004 (0.8768 ± 0.0039), 0.9923 (0.8861), 0.9898 (0.8880), and 0.8222 (0.7232) under five-fold CV, Global Leave-One-Out CV (LOOCV), miRNA-Fixed Local LOOCV, and SM-Fixed Local LOOCV, respectively, which are a significant improvement compared with previous models. In the first type of case study, 9 (33) among the top 20 (100) associations predicted by AMCSMMA are confirmed by the published experimental literature. In the second type of case study to the SMs 5-FU and 5-Aza-2’-deoxycytidine, 20 (34) and 16 (26) among the top 20 (50) associations are, respectively, verified by published references. In conclusion, AMCSMMA demonstrates superior accuracy and reliability in predicting potential SM–miRNA associations. It can be used for screening SM–miRNA samples with high association probabilities for complex biological experiments, thus significantly reducing the time and financial cost of discovering new SM–miRNA associations. This paper’s significant contributions are summarized as follows:
We integrate a variety of SM/miRNA similarities and consider the adjacency matrix of the constructed SM–miRNA heterogeneous network as the target matrix, which can not only effectively utilize the integrated similarity to improve the prediction accuracy but also enhance its information content as the iteration progresses.
We utilize the truncated nuclear norm regularization as the strategy to approximate the rank function, which not only achieves the rank minimization more accurately, robustly, and efficiently but also increases the adaptability to different datasets.
We design an effective two-step iterative scheme to solve the optimization problem. In order to solve the convex sub-problem in the second step, we introduce the Alternating Directional Multiplier Method (ADMM).
3. Results
3.1. Validation Experiment A
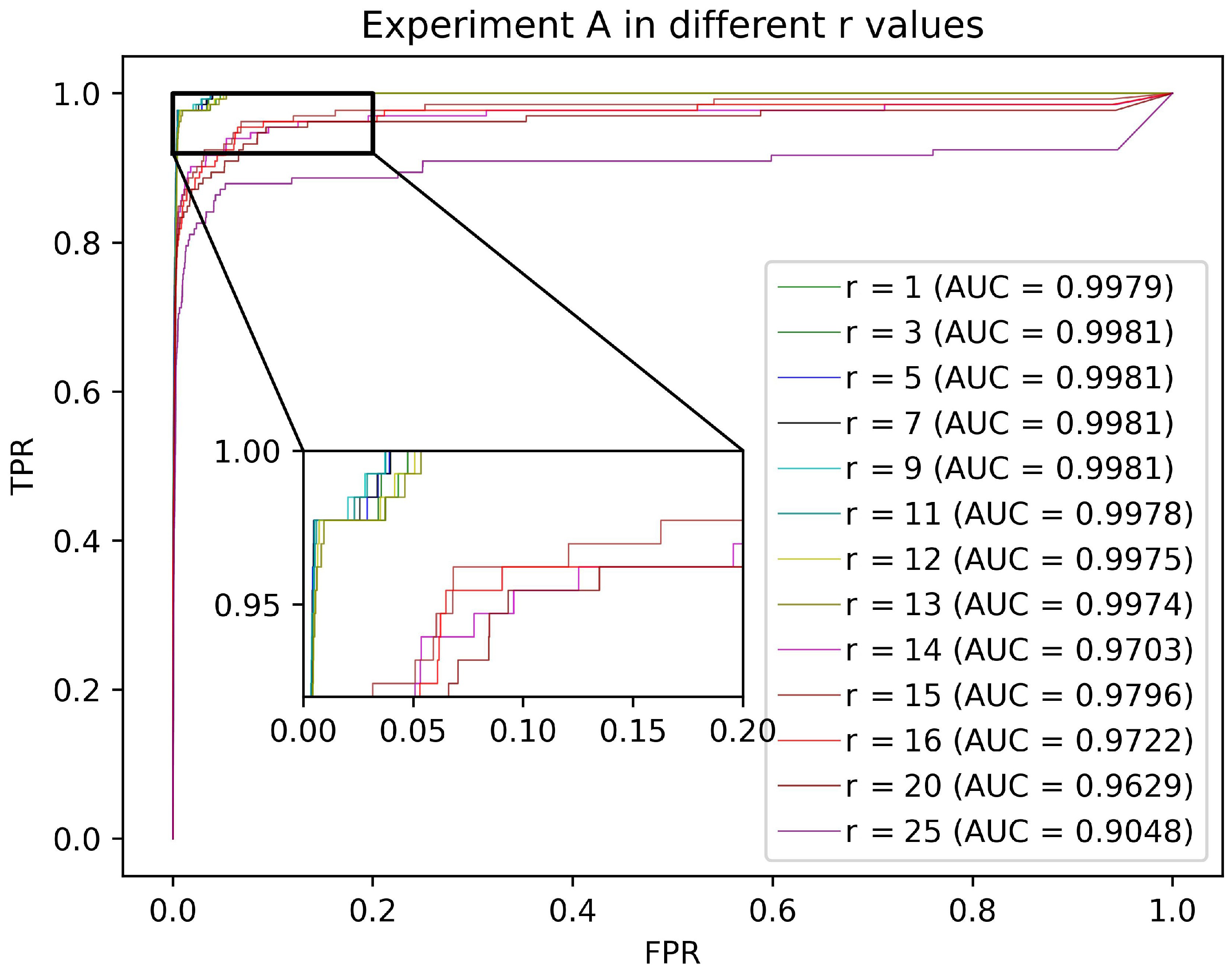
In this section, we design Validation Experiment A to quantitatively analyze the effect of the truncated position r on the predictive performance of AMCSMMA. Specifically, all confirmed associations in Dataset 1 are regarded as the training samples, all verified associations in Dataset 3 are treated as the testing samples, and all SM–miRNA pairs in Dataset 1 that are neither part of the training set nor the testing set are considered as the candidate samples.
Under specific , we conduct AMCSMMA only utilizing the training samples to recover the SM–miRNA association matrix with missing values. Then, the association scores of the candidate samples and the testing samples are extracted and arranged in descending order to calculate the False Positive Rate (FPR, 1-specificity) and the True Positive Rate (TPR, sensitivity) at a specific threshold. Furthermore, we set the FPR as the abscissa and the TPR as the ordinate and plot the Receiver Operating Characteristic (ROC) curves based on different thresholds.
The AUC between 0 and 1 is the area under the ROC curve, and the larger the numerical value is, the better the predictive performance of the model. According to
Figure 2, we find that AMCSMMA achieved excellent and stable performance when
. With the increase of r value in [1, 13], the computational complexity increased, whereas the prediction accuracy improved weakly. Considering that the adjustable target rank can increase the adaptability of the model to different datasets, we finally set
. The AUC reached 0.9981 at the optimal parameters, which strongly demonstrates the superiority of our model in predicting potential SM–miRNA associations.
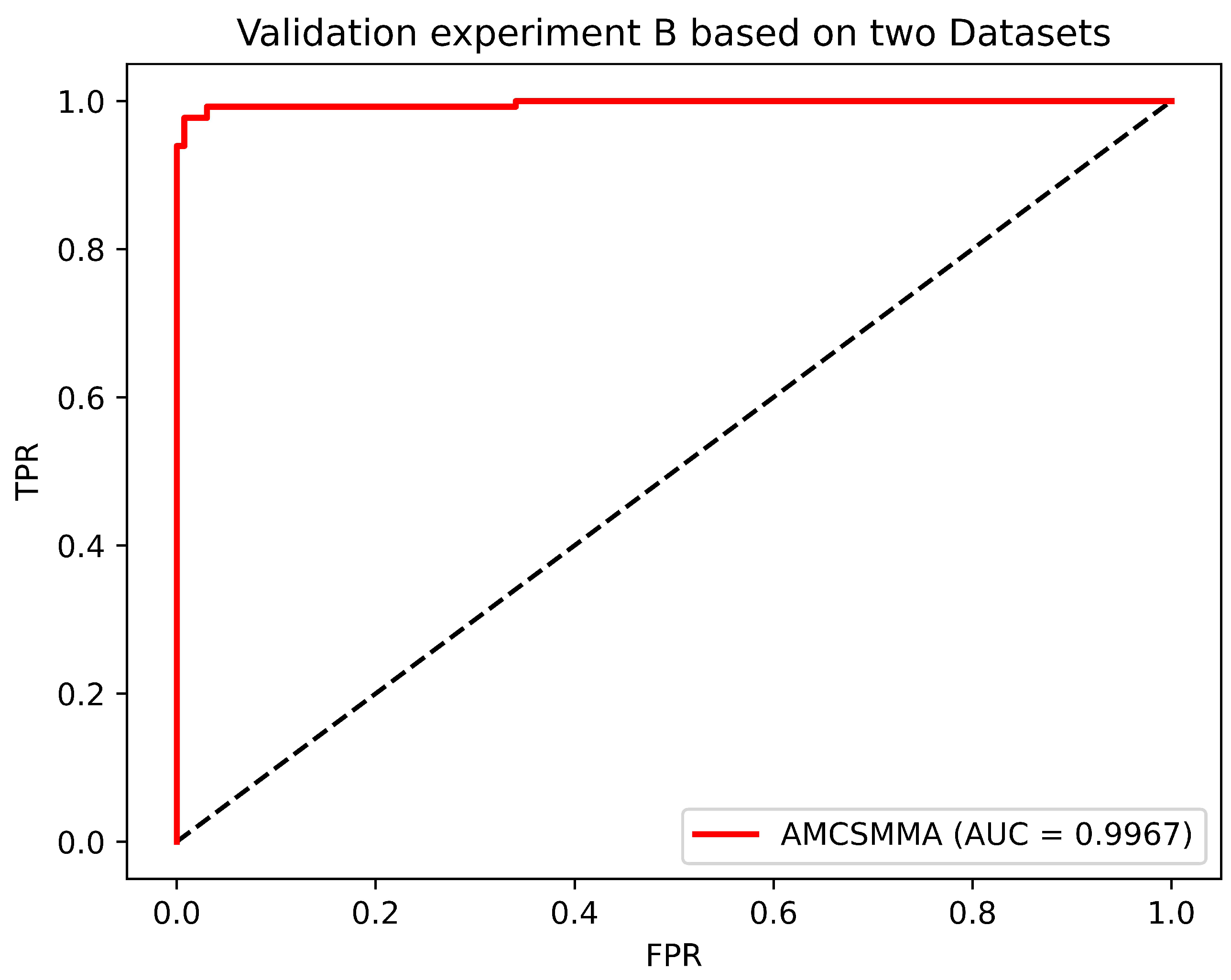
3.2. Validation Experiment B
To comprehensively evaluate the predictive performance of our model, we design Validation Experiment B, in which 664 confirmed SM–miRNA associations in Dataset 1 are utilized as the training samples, and 132 verified SM–miRNA associations in Dataset 3 are assembled into the positive testing set. Then, we randomly select 132 unknown SM–miRNA associations from Dataset 1 to form the negative testing set. The intersection set of the training set, the positive testing set, and the negative testing set is an empty set.
In addition, the AUC, five additional metrics are introduced, which include the Precision, Recall, F1 Score, Accuracy, and MCC. We calculate the above metrics based on three thresholds that maximize the F1 Score, the Accuracy, and the MCC. Considering the fluctuation of experimental results caused by randomly selecting the negative testing samples, we repeat the above procedure 100 times and consider the average value as the final result. It can be seen from
Table 3 and
Figure 3 that all metrics achieved a significant result.
3.3. Four Cross-Validation Experiments
Based on Dataset 1 and Dataset 2 separately, we implemented five-fold cross-validation (CV), Global Leave-One-Out CV (LOOCV), miRNA-Fixed Local LOOCV, and SM-Fixed Local LOOCV to further validate the predictive performance of AMCSMMA. At the same time, we likewise applied the above four CVs to other association predictive models.
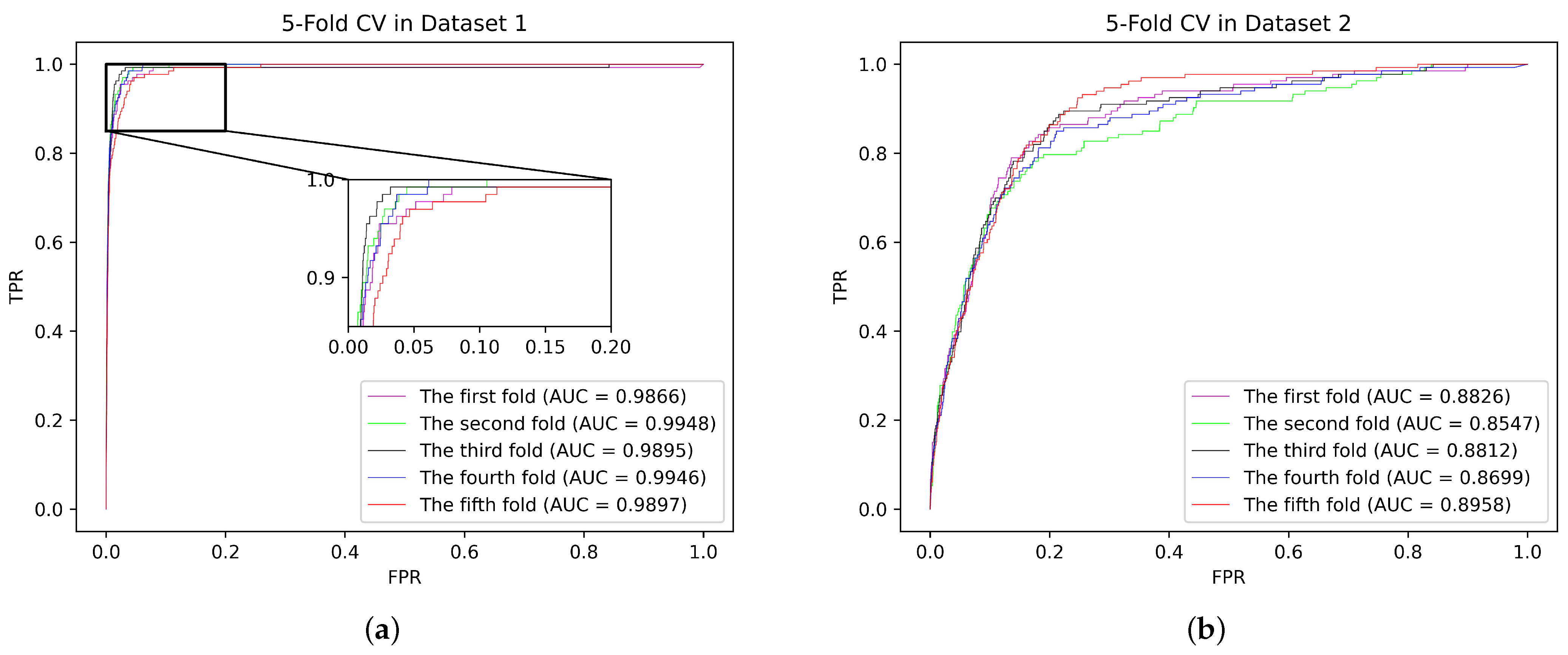
In the five-fold CV, all the confirmed SM–miRNA associations (664 items) were randomly divided into five parts, of which one part incorporated 132 items and each remaining part included 133 items. Specifically, we alternately utilized one part as the testing set, and the remaining four parts were fused as the training set. Additionally, all the unknown SM–miRNA associations were assembled in the candidate set. In each fold, only utilizing the training samples, we conducted AMCSMMA to recover the SM–miRNA association matrix. Likewise as in Validation Experiment A, the association scores of the testing and candidate samples were integrated into a descending sequence.
Then, we plotted the ROC curve and derived the AUC value under this fold. After five folds, the average AUC value was regarded as the result of one five-fold CV. It is worth noting that we repeated the five-fold CV 100 times and took the average AUC value as the final result, which insulates the validation result against the randomness of sample partitioning. Additionally, we calculated the Standard Deviation (SD) value that can reflect the robustness of the model. Finally, the AUC±SD of AMCSMMA under five-fold CV reached 0.9910 ± 0.0004 and 0.8768 ± 0.0039 based on Datasets 1 and 2, respectively.
From
Table 4, we observe that AMCSMMA achieved a higher AUC and a lower SD than did the compared models based on both datasets, which indicates that it has superior predictive performance and robustness.
Figure 4 shows the ROC curves of each fold in one five-fold CV based on two datasets and the areas under the curves.
In Global LOOCV, each verified SM–miRNA association was sequentially selected as the testing sample, and the remaining 663 confirmed associations were considered as the training samples. Additionally, all unknown SM–miRNA associations were treated as the candidate samples.
Similarly, we calculated the AUC values successively under 664 folds according to the association scores of the testing and candidate samples and regarded the average AUC as the result. From
Table 4, we discover that the AUC of AMCSMMA under Global LOOCV reached 0.9923 (0.8861) based on Dataset 1 (Dataset 2), which exceeds all other models proposed in recent years and once again demonstrates the superior predictive performance of our model.
In miRNA-Fixed Local LOOCV and SM-Fixed Local LOOCV, the testing and training samples were selected in the same way as in Global LOOCV. Nevertheless, the candidate set in miRNA/SM-Fixed Local LOOCV only consisted of the unknown SM–miRNA associations that have the same miRNA/SM with the testing sample in each fold. After several computational steps, the AUC of AMCSMMA reached 0.9898 (0.8880) based on Dataset 1 (Dataset 2) under miRNA-Fixed Local LOOCV, which surpasses all comparative models. The AUC reached 0.8222 (0.7232) based on Dataset 1 (Dataset 2) under SM-Fixed Local LOOCV, which is superior to the other four models (TLHNSMMA, GISMMA, SLHGISMMA, and SMiR-NBI). The DCMF achieved the best performance because it was able to obtain the exact SM feature matrix.
As shown in
Table 4, AMCSMMA achieved better performance based on Dataset 1 than on Dataset 2 in cross-validation experiments. The reason for this is that Datasets 1 and 2 provide the same positive samples (divided into training and testing samples), but Dataset 1 provides a much larger number of candidate samples compared with Dataset 2. Since these additional candidate samples contain SMs/miRNAs that have no known associations with miRNAs/SMs, they have relatively low association scores compared to the testing samples, resulting in a higher AUC value based on Dataset 1 than on Dataset 2. Therefore, we expect that the accuracy of AMCSMMA will improve as more SMs and miRNAs are added to the dataset.
3.4. Case Studies
3.4.1. The First Type of Case Study
In this section, we initially utilize AMCSMMA to obtain the predictive scores of all unknown SM–miRNA associations in Dataset 1. Subsequently, we count the number of associations confirmed by published literature in PubMed. Finally, 9 (33) among the top 20 (100) associations can be confirmed.
Table 5 lists the top 20 associations and the literature evidence (PubMed ID).
Specifically, Khorrami et al. [
45] identified that miR-146a is overexpressed in a colon cancer cell line (HT-29), which can increase its resistance to 5-FU and irinotecan, thereby diminishing the prognostic effect of chemotherapy. Additionally, Zhang et al. [
46] revealed that CYP11A1 and CYP19A1 expression in human CCs, and the resulting production of progesterone and estradiol, are transcriptionally down-regulated by miR-320a deficiency. Moreover, the colorectal cancer hallmark (CXCL12) is able to induce miR-125 upregulation and generate the chemotherapy drugs 5-FU resistance [
47].
We implement this type of case study on other comparative models. From
Table 6, our model achieves the best performance.
3.4.2. The Second Type of Case Study
To explore the applicability of AMCSMMA to new SMs, we conducted the second type of case study to two SMs, 5-FU and 5-Aza-2’-deoxycytidine based on Dataset 1. In detail, we first removed all verified associations related to the specific SM. Then, a descending sequence consisting of association scores between the specific SM and all miRNAs was obtained. We counted the number of associations confirmed by the SM2miR database [
27] and published references. Finally, in the second type to 5-FU, 20 (34) among the top 20 (50) associations were confirmed as shown in
Table 7.
Specifically, the sensitivity of 5-FU was significantly correlated with the antitumor effect, and overexpression of miR-329 and let-7c enhanced the sensitivity of 5-FU by affecting the apoptotic pathway, thus enhancing the antitumor effect [
48,
49]. In another study, Wang et al. [
50] found that 5-FU was abnormally sensitive to MCF-7 cells due to its negative regulation on Bcl-xl expression via let-7b. Additionally, Bamodu et al. [
51] concluded that the SOD2-enhanced 5-FU chemoresistance of colorectal cancer cells was inhibited by inducing the re-expression of hsa-miR-324. Furthermore, Han et al. [
52] discovered that miR-874 can reduce the resistance of colorectal cancer cells to 5-FU.
In the second type to 5-Aza-2’-deoxycytidine, 16 (26) of the top 20 (50) associations were confirmed as shown in
Table 8. Particularly, Liu et al. [
53] found that the demethylation agent 5-Aza-2’-deoxycytidine inhibited the proliferation of esophageal cancer cells by increasing the expression of miR-203a. Moreover, the expression of miR-19b and let-7b increased in gastric cancer cells after 5-Aza-2’-deoxycytidine treatment [
54,
55]. In addition, Sun et al. [
56] found that hypermethylation of the promoter region in gastrointestinal cancer cell lines correlated with the expression of miR-148a in gastric cancer, and thus treatment with the demethylation agent 5-Aza-2’-deoxycytidine can be performed.
Furthermore, we conducted the second type of case study on BNNRSMMA and DCMF, which are both heuristic algorithms based on matrix completion. As shown in
Table 9, our model achieved the best performance except in Number D.
In conclusion, the above experimental results demonstrate that AMCSMMA is an excellent model with superior predictive performance and high robustness in predicting potential SM–miRNA associations, which can provide guidance for complex and expensive biological experiments and accelerate the discovery of new SM–miRNA associations, thus facilitating drug development and disease treatment.
4. Discussion
In recent years, an increasing number of studies have shown that the abnormal expression of miRNAs is closely related to a variety of physiological and pathological processes, including cancer, cardiovascular diseases, and metabolic diseases [
13,
57]. As a result, targeting and modulating miRNAs with small molecule (SM) drugs has become a significant modality for clinical treatment.
Given the complexity and expense of developing new SMs, it is extremely difficult to develop specific SMs for each dysregulated miRNA. Therefore, exploring potential associations between known SMs and miRNAs is both significant and urgent in drug development and disease treatment. Since confirming SM–miRNA associations through biological experiments is time-consuming and expensive, more effective predictive approaches need to be proposed for identifying the SM–miRNA associations with high association probabilities, which can provide guidance for biological experiments and discover potential SM–miRNA associations more cost-effectively.
In this paper, we proposed a more accurate predictive model based on the truncated nuclear norm, called AMCSMMA. After determining the optimal parameter values, the results of Validation Experiment A, four cross-validation experiments, Validation Experiment B, and two types of case studies indicated that AMCSMMA had superior prediction accuracy and high robustness. The reasons for this are discussed in the following.
All the known SM–miRNA associations were acquired from the SM2miR v1.0 database [
27] and the published experimental literature, which are extremely reliable.
We constructed the SM–miRNA heterogeneous network and defined its adjacency matrix as the target matrix. This not only well utilized similarity information but also enriched it as the iteration progressed.
Unlike the nuclear norm regularization, the truncated nuclear norm regularization only minimized the sum of partial singular values, which not only made the result matrix more closely approximate the true solution but also improved the adaptability to different datasets.
We designed an effective two-step iterative scheme to solve the optimization problem.
Although the advancement of AMCSMMA in predicting potential SM–miRNA associations enables it to provide reliable guidance for biological experiments, the model still has some limitations. For instance, the small number of known SM–miRNA associations greatly restricts the prediction accuracy of our model. Moreover, the biological data closely related to SM or miRNA, such as lncRNA and the circRNA, can be introduced to construct heterogeneous networks with more information to improve the prediction accuracy. Furthermore, the work of Yu et al. [
58] inspired the idea that deep-learning-based approaches may be able to achieve good results. Due to the multiple utilization of the SVD algorithm, our model requires a relatively high time cost, which will be the focus of our future research.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}