
EZH2 Methyltransferase Regulates Neuroinflammation and Neuropathic Pain
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. EZH2 Biological Function and Its Inhibitors
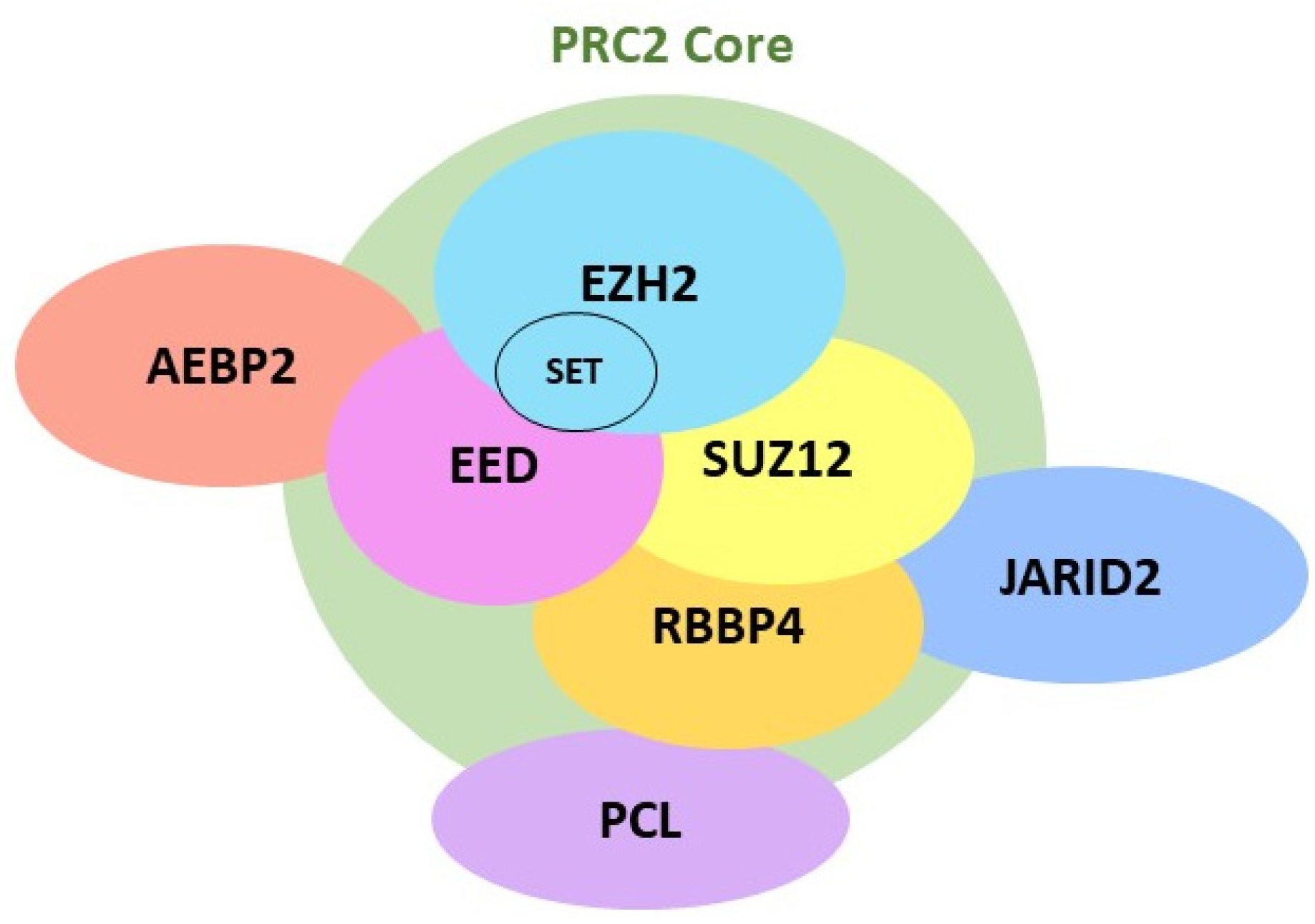
2.1. EZH2 Biological Function
2.2. EZH2 Inhibitors
3. Regulation of Neuroinflammation and Neuropathic Pain by EZH2
3.1. Temporal Correlation between Enhanced EZH2 Protein Expression and Neuroinflammation Induced by Nerve Injury
3.2. Inhibition of EZH2 Ameliorates Neuroinflammation and Neuropathic Pain
4. Upstream Signaling Molecules Regulating EZH2 Protein Expression and Function
4.1. TLR4
4.2. CGRP
4.3. miR-124-3p
4.4. miR-378
4.5. Lncenc1
4.6. MALAT1
5. Downstream Signaling Molecules Used by EZH2 to Regulate Neuroinflammation and Neuropathic Pain
5.1. miR-146a-5p/HIF-1α
5.2. miR-29b-3p/MMP-2
5.3. SOCS3
5.4. Nrf2
5.5. BAI1
6. Mechanisms Used by Glia to Cause Aberrant Neuronal Activity along the Pain Signaling Pathway in Neuropathic Pain
7. Concluding Remarks
8. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.A. The Known Biology of Neuropathic Pain and Its Relevance to Pain Management. Can. J. Neurol. Sci. 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, D.S.M.; Nassini, R.; Geppetti, P.; De Logu, F. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin. Ther. Targets 2020, 24, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Weng, H.R. Endogenous interleukin-1beta in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic N-methyl-D-aspartic acid receptors. J. Biol. Chem. 2013, 288, 30544–30557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, H.; Weng, H.-R. Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. J. Neurophysiol. 2010, 103, 2570–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, H.; Zhang, H.; Weng, H.R. Minocycline prevents impaired glial glutamate uptake in the spinal sensory synapses of neuropathic rats. Neuroscience 2010, 170, 901–912. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Penas, C.; Navarro, X. Epigenetic Modifications Associated to Neuroinflammation and Neuropathic Pain After Neural Trauma. Front. Cell Neurosci. 2018, 12, 158. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.; Weng, H.-R. EZH2 regulates spinal neuroinflammation in rats with neuropathic pain. Neuroscience 2017, 349, 106–117. [Google Scholar] [CrossRef] [Green Version]
- An, Q.; Sun, C.; Li, R.; Chen, S.; Gu, X.; An, S.; Wang, Z. Calcitonin gene-related peptide regulates spinal microglial activation through the histone H3 lysine 27 trimethylation via enhancer of zeste homolog-2 in rats with neuropathic pain. J. Neuroinflammation 2021, 18, 117. [Google Scholar] [CrossRef]
- Meng, X.-L.; Fu, P.; Wang, L.; Yang, X.; Hong, G.; Zhao, X.; Lao, J. Increased EZH2 Levels in Anterior Cingulate Cortex Microglia Aggravate Neuropathic Pain by Inhibiting Autophagy Following Brachial Plexus Avulsion in Rats. Neurosci. Bull. 2020, 36, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gu, X.; Li, R.; An, S.; Wang, Z. Genome-wide Analysis of Histone H3 Lysine 27 Trimethylation Profiles in Sciatic Nerve of Chronic Constriction Injury Rats. Neurochem. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, X.; Zhao, G.; Ma, Y.; Zeng, G. LncRNA embryonic stem cells expressed 1 (Lncenc1) is identified as a novel regulator in neuropathic pain by interacting with EZH2 and downregulating the expression of Bai1 in mouse microglia. Exp. Cell Res. 2021, 399, 112435. [Google Scholar] [CrossRef]
- Ni, S.; Yang, B.; Xia, L.; Zhang, H. EZH2 Mediates miR-146a-5p/HIF-1alpha to Alleviate Inflammation and Glycolysis after Acute Spinal Cord Injury. Mediat. Inflamm. 2021, 2021, 5591582. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zeng, X.; Zhang, L.; Wang, L.; Shen, L.-L.; Hou, Y.-Y.; Zhou, F.; Zhang, X. Overexpression of miR-378 Alleviates Chronic Sciatic Nerve Injury by Targeting EZH2. Neurochem. Res. 2021, 46, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Torres-Perez, J.V.; Irfan, J.; Febrianto, M.R.; Di Giovanni, S.; Nagy, I. Histone post-translational modifications as potential therapeutic targets for pain management. Trends Pharmacol. Sci. 2021, 42, 897–911. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an easy way to harmonize: A review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin. Epigenetics 2021, 13, 62. [Google Scholar] [CrossRef]
- Lim, H.J.; Kim, M. EZH2 as a Potential Target for NAFLD Therapy. Int. J. Mol. Sci. 2020, 21, 8617. [Google Scholar] [CrossRef]
- Liu, Z.; Jia, Y.; Guo, Y.; Wang, H.; Fu, R. Role of EZH2 in bone marrow mesenchymal stem cells and immune–cancer interactions. Crit. Rev. Oncol. Hematol. 2021, 169, 103547. [Google Scholar] [CrossRef]
- Su, I.H.; Dobenecker, M.W.; Dickinson, E.; Oser, M.; Basavaraj, A.; Marqueron, R.; Viale, A.; Reinberg, D.; Wülfing, C.; Tarakhovsky, A. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell 2005, 121, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poepsel, S.; Kasinath, V.; Nogales, E. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol. 2018, 25, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef]
- Kipp, D.R.; Quinn, C.M.; Fortin, P.D. Enzyme-dependent lysine deprotonation in EZH2 catalysis. Biochemistry 2013, 52, 6866–6878. [Google Scholar] [CrossRef] [PubMed]
- Højfeldt, J.W.; Hedehus, L.; Laugesen, A.; Tatar, T.; Wiehle, L.; Helin, K. Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol. Cell 2019, 76, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.D.; Li, J.; Wong, J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell Biol. 2005, 25, 2525–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirabayashi, Y.; Suzki, N.; Tsuboi, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Koseki, H.; Vidal, M.; Gotoh, Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009, 63, 600–613. [Google Scholar] [CrossRef] [Green Version]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Gu, X.; Su, I.H.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, K.; Liu, M.; Zhang, H.; Guo, M. EZH2: Its regulation and roles in immune disturbance of SLE. Front. Pharmacol. 2022, 13, 1002741. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.T.; Lin, C.A. Role of the histone methyltransferases Ezh2 and Suv4-20h1/Suv4-20h2 in neurogenesis. Neural Regen. Res. 2023, 18, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, G.; Zhang, K.; Cao, J.; Zhao, H.; Cong, Y.; Qiao, G. Integrated transcriptome and network analysis identifies EZH2/CCNB1/PPARG as prognostic factors in breast cancer. Front. Genet. 2022, 13, 1117081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bui, T.; Zhang, Y. The pleiotropic roles of EZH2 in T-cell immunity and immunotherapy. Int. J. Hematol. 2022, 116, 837–845. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, S.; Wang, Z.; Huang, J.; Xu, L.; Tang, X.; Wan, Y.Y.; Li, Q.-J.; Symonds, A.L.J.; Long, H.; et al. Targeting EZH2 histone methyltransferase activity alleviates experimental intestinal inflammation. Nat. Commun. 2019, 10, 2427. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.; Liu, W.; Liu, Y.; Xu, J. Polycomb-mediated loss of microRNA let-7c determines inflammatory macrophage polarization via PAK1-dependent NF-kappaB pathway. Cell Death Differ. 2015, 22, 287–297. [Google Scholar] [CrossRef]
- Yuan, J.-L.; Yin, C.-Y.; Li, Y.-Z.; Song, S.; Fang, G.-J.; Wang, Q.-S. EZH2 as an Epigenetic Regulator of Cardiovascular Development and Diseases. J. Cardiovasc. Pharmacol. 2021, 78, 192–201. [Google Scholar] [CrossRef]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [Green Version]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [Green Version]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’Ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Södersten, E.; Feyder, M.; Lerdrup, M.; Gomes, A.-L.; Kryh, H.; Spigolon, G.; Caboche, J.; Fisone, G.; Hansen, K. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLOS Genet. 2014, 10, e1004574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Fang, Y.; Kang, R.; Lenahan, C.; Gamdzyk, M.; Zhang, Z.; Okada, T.; Tang, J.; Chen, S.; Zhang, J.H. Inhibition of EZH2 (Enhancer of Zeste Homolog 2) Attenuates Neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-kappaB (Trimethylation of Histone 3 Lysine 27/Suppressor of Cytokine Signaling 3/Tumor Necrosis Factor Receptor Family 6/Nuclear Factor-kappaB) in a Rat Model of Subarachnoid Hemorrhage. Stroke 2020, 51, 3320–3331. [Google Scholar] [PubMed]
- Jiang, X.; Lim, C.Z.; Li, Z.; Lee, P.L.; Yatim, S.M.; Guan, P.; Li, J.; Zhou, J.; Pan, J.; Chng, W.-J.; et al. Functional Characterization of D9, a Novel Deazaneplanocin A (DZNep) Analog, in Targeting Acute Myeloid Leukemia (AML). PLoS ONE 2015, 10, e0122983. [Google Scholar] [CrossRef]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y. Targeting histone methyltransferase EZH2 as cancer treatment. J. Biochem. 2014, 156, 249–257. [Google Scholar] [CrossRef] [PubMed]
- First EZH2 Inhibitor Approved-for Rare Sarcoma. Cancer Discov. 2020, 10, 333–334. [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, Q.; Xie, L.; Lei, S. Histone methyltransferase enhancer of zeste 2 polycomb repressive complex 2 subunit exacerbates inflammation in depression rats by modulating microglia polarization. Bioengineered 2022, 13, 5509–5524. [Google Scholar] [CrossRef]
- Lehnardt, S.; Lachance, C.; Patrizi, S.; Lefebvre, S.; Follett, P.L.; Jensen, F.E.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 2002, 22, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, X.; Ke, A. High-mobility group box-1 induces mechanical pain hypersensitivity through astrocytic connexin 43 via the toll-like receptor-4/JNK signaling pathway. Synapse 2020, 75, e22184. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, R.; Tsubota, M.; Sekiguchi, F.; Kawabata, A. Role of high-mobility group box 1 and its modulation by thrombomodulin/thrombin axis in neuropathic and inflammatory pain. Br. J. Pharmacol. 2020, 178, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.R.; Ramos, K.M.; Loram, L.C.; Wieseler, J.; Sholar, P.W.; Kearney, J.J.; Lewis, M.T.; Crysdale, N.Y.; Zhang, Y.; Harrison, J.A.; et al. Evidence for a role of heat shock protein-90 in toll like receptor 4 mediated pain enhancement in rats. Neuroscience 2009, 164, 1821–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, B.; Xu, A.; Liang, X.; Xu, T.; Jin, H.; Xie, Y.; Wang, R.; Liu, X.; Gao, X.; et al. NF-kappaB and AP-1 are required for the lipopolysaccharide-induced expression of MCP-1, CXCL1, and Cx43 in cultured rat dorsal spinal cord astrocytes. Front. Mol. Neurosci. 2022, 15, 859558. [Google Scholar] [CrossRef]
- Bettoni, I.; Comelli, F.; Rossini, C.; Granucci, F.; Giagnoni, G.; Peri, F.; Costa, B. Glial TLR4 receptor as new target to treat neuropathic pain: Efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 2008, 56, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Tanga, F.Y.; Nutile-McMenemy, N.; DeLeo, J.A. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. USA 2005, 102, 5856–5861. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.-X.; Bian, J.-J.; Miao, X.-R.; Huang, S.-D.; Xu, X.-W.; Gong, D.-J.; Sun, Y.-M.; Lu, Z.-J.; Yu, W.-F. Intrathecal siRNA against Toll-like receptor 4 reduces nociception in a rat model of neuropathic pain. Int. J. Med. Sci. 2010, 7, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, X.-W.; Feng, X.-M.; Zhou, W.; Wang, Y.-Q.; Mao-Ying, Q.-L. Stage-dependent anti-allodynic effects of intrathecal Toll-like receptor 4 antagonists in a rat model of cancer induced bone pain. J. Physiol. Sci. 2013, 63, 203–209. [Google Scholar] [CrossRef]
- Okui, T.; Hiasa, M.; Ryumon, S.; Ono, K.; Kunisada, Y.; Ibaragi, S.; Sasaki, A.; Roodman, G.D.; White, F.A.; Yoneda, T. The HMGB1/RAGE axis induces bone pain associated with colonization of 4T1 mouse breast cancer in bone. J. Bone Oncol. 2021, 26, 100330. [Google Scholar] [CrossRef]
- Sorge, R.E.; LaCroix-Fralish, M.L.; Tuttle, A.H.; Sotocinal, S.G.; Austin, J.-S.; Ritchie, J.; Chanda, M.L.; Graham, A.C.; Topham, L.; Beggs, S.; et al. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J. Neurosci. 2011, 31, 15450–15454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Li, F.; Maixner, D.W.; Yadav, R.; Gao, M.; Ali, M.W.; Hooks, S.B.; Weng, H.-R. Interleukin-1beta released by microglia initiates the enhanced glutamatergic activity in the spinal dorsal horn during paclitaxel-associated acute pain syndrome. Glia 2019, 67, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Maixner, D.W.; Yadav, R.; Gao, M.; Li, P.; Bartlett, M.G.; Weng, H.-R. Paclitaxel induces acute pain via directly activating toll like receptor 4. Mol. Pain 2015, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, M.F.; Hügle, T.; Morlion, B.; Koltzenburg, M.; Chapman, V.; MaassenVanDenBrink, A.; Lane, N.E.; Perrot, S.; Zieglgänsberger, W. Neurogenic inflammation as a novel treatment target for chronic pain syndromes. Exp. Neurol. 2022, 356, 114108. [Google Scholar] [CrossRef] [PubMed]
- Schou, W.S.; Ashina, S.; Amin, F.M.; Goadsby, P.J.; Ashina, M. Calcitonin gene-related peptide and pain: A systematic review. J. Headache Pain 2017, 18, 34. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xue, Y.; Lu, Q.; Shi, Y.; Tang, W.; Wang, D. Adrenomedullin is an Important Pathological Mediator in Progression of Chronic Neuropathic Pain. Front. Biosci. 2022, 27, 220. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; An, Q.; Li, R.; Chen, S.; Gu, X.; An, S.; Wang, Z. Calcitonin gene-related peptide induces the histone H3 lysine 9 acetylation in astrocytes associated with neuroinflammation in rats with neuropathic pain. CNS Neurosci. Ther. 2021, 27, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Presto, P.; Neugebauer, V. Sex Differences in CGRP Regulation and Function in the Amygdala in a Rat Model of Neuropathic Pain. Front. Mol. Neurosci. 2022, 15, 262. [Google Scholar] [CrossRef]
- Xiong, W.; Huang, L.; Shen, Y.; Guan, S.; He, L.; Tong, Z.; Tan, M.; Liu, L.; Gao, Y. Effects of lncRNA uc.48+ siRNA on the release of CGRP in the spinal cords of rats with diabetic neuropathic pain. Int. J. Clin. Exp. Pathol. 2017, 10, 9960–9969. [Google Scholar]
- Zhang, Y.; Liu, H.L.; An, L.J.; Li, L.; Wei, M.; Ge, D.J.; Su, Z. miR-124-3p attenuates neuropathic pain induced by chronic sciatic nerve injury in rats via targeting EZH2. J. Cell. Biochem. 2019, 120, 5747–5755. [Google Scholar] [CrossRef]
- Zhang, W.-C.; Liu, J.; Xu, X.; Wang, G. The role of microRNAs in lung cancer progression. Med Oncol. 2013, 30, 675. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118, Correction in 2021, 22, 159. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhu, M.; Lv, P.; Cheng, L.; Wang, Q.; Tian, P.; Yan, Z.; Wen, B. The Long Noncoding RNA Lncenc1 Maintains Naive States of Mouse ESCs by Promoting the Glycolysis Pathway. Stem Cell Rep. 2018, 11, 741–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, C.; Ding, H. LncRNA MALAT1 promotes neuropathic pain progression through the miR-154-5p/AQP9 axis in CCI rat models. Mol. Med. Rep. 2020, 21, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-L.; Liu, J.-Y.; Wang, F.; Jing, X. Suppression of MALAT1 ameliorates chronic constriction injury-induced neuropathic pain in rats via modulating miR-206 and ZEB2. J. Cell. Physiol. 2019, 234, 15647–15653. [Google Scholar] [CrossRef]
- Cai, L.-J.; Tu, L.; Huang, X.-M.; Huang, J.; Qiu, N.; Xie, G.-H.; Liao, J.-X.; Du, W.; Zhang, Y.-Y.; Tian, J.-Y. LncRNA MALAT1 facilitates inflammasome activation via epigenetic suppression of Nrf2 in Parkinson’s disease. Mol. Brain 2020, 13, 130. [Google Scholar] [CrossRef]
- Alam, H.; Gu, B.; Lee, M.G. Histone methylation modifiers in cellular signaling pathways. Cell. Mol. Life Sci. 2015, 72, 4577–4592. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Mao, C.; Liu, J.; Yu, T.; et al. Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3. J. Exp. Med. 2018, 215, 1365–1382. [Google Scholar] [CrossRef]
- Lu, Y.; Cao, D.-L.; Jiang, B.-C.; Yang, T.; Gao, Y.-J. MicroRNA-146a-5p attenuates neuropathic pain via suppressing TRAF6 signaling in the spinal cord. Brain Behav. Immun. 2015, 49, 119–129. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, Y.; Xu, Z.-Z.; Wang, X.; Park, J.Y.; Zhuang, Z.-Y.; Tan, P.-H.; Gao, Y.-J.; Roy, K.; Corfas, G.; Lo, E.H.; et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 2008, 14, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Wang, C.; Zhang, L.; Song, L.; Chen, Y.; Liu, B.; Liu, W.T.; Hu, L.; Pan, Y. Procyanidins attenuate neuropathic pain by suppressing matrix metalloproteinase-9/2. J. Neuroinflammation 2018, 15, 187. [Google Scholar] [CrossRef]
- Li, L.; Zhao, G. Downregulation of microRNA-218 relieves neuropathic pain by regulating suppressor of cytokine signaling 3. Int. J. Mol. Med. 2016, 37, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, E.; Mauborgne, A.; Mallet, J.; Desclaux, M.; Pohl, M. SOCS3-mediated blockade of JAK/STAT3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. J. Neurosci. 2010, 30, 5754–5766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.-Q.; Mei, W.; Tian, X.-B.; Tian, Y.-K.; Liu, D.-Q.; Ye, D.-W. The therapeutic potential of Nrf2 inducers in chronic pain: Evidence from preclinical studies. Pharmacol. Ther. 2021, 225, 107846. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef]
- Martin, S.J.; Reutelingsperger, C.P.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.; LaFace, D.M.; Green, D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [Green Version]
- Mazaheri, F.; Breus, O.; Durdu, S.; Haas, P.; Wittbrodt, J.; Gilmour, D.; Peri, F. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 2014, 5, 4046. [Google Scholar] [CrossRef] [Green Version]
- Awan, M.U.; Deng, Y. Role of autophagy and its significance in cellular homeostasis. Appl. Microbiol. Biotechnol. 2014, 98, 5319–5328. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular selfdigestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Sakamoto, K. Ethyl pyruvate protects SHSY5Y cells against 6-hydroxydopamine-induced neurotoxicity by upregulating autophagy. PLoS ONE 2023, 18, e0281957. [Google Scholar] [CrossRef] [PubMed]
- Tchekalarova, J.; Tzoneva, R. Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin. Int. J. Mol. Sci. 2023, 24, 3022. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Li, Z.; Liang, G.; Zhou, R.; Zheng, X.; Tao, R.; Huo, Q.; Su, C.; Li, M.; Xu, N.; et al. TNEA therapy promotes the autophagic degradation of NLRP3 inflammasome in a transgenic mouse model of Alzheimer’s disease via TFEB/TFE3 activation. J. Neuroinflammation 2023, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, S.-Y.; Wang, Q.-G.; Xu, Z.-H.; Peng, Q.; Chen, S.-Y.; Zhu, L.; Zhang, Y.-D.; Duan, R. AVE 0991 Suppresses Astrocyte-Mediated Neuroinflammation of Alzheimer’s Disease by Enhancing Autophagy. J. Inflamm. Res. 2023, 16, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Lu, J.-H. Targeting chaperone-mediated autophagy for Parkinson’s disease therapy. Neural Regen. Res. 2023, 18, 1723–1724. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Ma, B.; Gao, N.; Jin, T.; Liu, X. Translocator Protein (TSPO) Alleviates Neuropathic Pain by Activating Spinal Autophagy and Nuclear SIRT1/PGC-1alpha Signaling in a Rat L5 SNL Model. J. Pain. Res. 2022, 15, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-F.; Lu, K.-T.; Hsu, J.-L.; Lee, C.-H.; Cheng, M.-Y.; Ro, L.-S. The Role of Autophagy and Apoptosis in Neuropathic Pain Formation. Int. J. Mol. Sci. 2022, 23, 2685. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Ji, R.-R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Jiang, E.; Gao, M.; Weng, H.-R. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J. Physiol. 2013, 591, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Zhang, L.; Cheng, J.K.; Ji, R.R. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 2008, 28, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Berta, T.; Park, C.-K.; Xu, Z.-Z.; Xie, R.-G.; Liu, T.; Lü, N.; Liu, Y.-C.; Ji, R.-R. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J. Clin. Investig. 2014, 124, 1173–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-J.; Zhang, L.; Samad, O.A.; Suter, M.R.; Yasuhiko, K.; Xu, Z.-Z.; Park, J.-Y.; Lind, A.-L.; Ma, Q.; Ji, R.-R. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 2009, 29, 4096–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, B.; Lim, G.; Mao, J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J. Neurosci. 2003, 23, 2899–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, H.; Weng, H.-R. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J. Neurophysiol. 2009, 101, 2041–2051. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Yadav, R.; Gao, M.; Weng, H.-R. Interleukin-1 beta enhances endocytosis of glial glutamate transporters in the spinal dorsal horn through activating protein kinase C. Glia 2014, 62, 1093–1109. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Zhang, H.; Weng, H.-R. Bidirectional neuron–glia interactions triggered by deficiency of glutamate uptake at spinal sensory synapses. J. Neurophysiol. 2010, 104, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Jiang, E.; Weng, H.-R. Activation of toll like receptor 4 attenuates GABA synthesis and postsynaptic GABA receptor activities in the spinal dorsal horn via releasing interleukin-1 beta. J. Neuroinflammation 2015, 12, 222. [Google Scholar] [CrossRef] [Green Version]
- Bonalume, V.; Caffino, L.; Castelnovo, L.F.; Faroni, A.; Giavarini, F.; Liu, S.; Caruso, D.; Schmelz, M.; Fumagalli, F.; Carr, R.W.; et al. Schwann Cell Autocrine and Paracrine Regulatory Mechanisms, Mediated by Allopregnanolone and BDNF, Modulate PKCepsilon in Peripheral Sensory Neurons. Cells 2020, 9, 1874. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Qu, L.; Wang, S.; Kim, M.; Bennett, D.; Ro, J.; Caterina, M.J.; Chung, M.-K. Phosphorylation of TRPV1 S801 Contributes to Modality-Specific Hyperalgesia in Mice. J. Neurosci. 2019, 39, 9954–9966. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Shao, J.; Cao, J.; Ren, X.; Cai, W.; Su, S.; George, S.; Tan, Z.; Zang, W.; Dong, T. Protein kinase C-alpha upregulates sodium channel Nav1.9 in nociceptive dorsal root ganglion neurons in an inflammatory arthritis pain model of rat. J. Cell Biochem. 2020, 121, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Toth, P.T.; White, F.A.; Miller, R.J. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J. Neurochem. 2008, 104, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippoldt, E.K.; Elmes, R.R.; McCoy, D.D.; Knowlton, W.M.; McKemy, D.D. Artemin, a glial cell line-derived neurotrophic factor family member, induces TRPM8-dependent cold pain. J. Neurosci. 2013, 33, 12543–12552. [Google Scholar] [CrossRef] [PubMed]
- Skoff, A.M.; Zhao, C.; Adler, J.E. Interleukin-1alpha regulates substance P expression and release in adult sensory neurons. Exp. Neurol. 2009, 217, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Botz, B.; Imreh, A.; Sándor, K.; Elekes, K.; Szolcsányi, J.; Reglődi, D.; Quinn, J.P.; Stewart, J.; Zimmer, A.; Hashimoto, H.; et al. Role of Pituitary Adenylate-Cyclase Activating Polypeptide and Tac1 gene derived tachykinins in sensory, motor and vascular functions under normal and neuropathic conditions. Peptides 2013, 43, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, S.; Alvarado-Vázquez, P.A.; Eisenach, J.C.; Romero-Sandoval, E.A.; Boada, M.D. Tachykinins modulate nociceptive responsiveness and sensitization: In vivo electrical characterization of primary sensory neurons in tachykinin knockout (Tac1 KO) mice. Mol. Pain 2019, 15. [Google Scholar] [CrossRef] [Green Version]
- Laumet, G.; Garriga, J.; Chen, S.-R.; Zhang, Y.; Li, D.-P.; Smith, T.M.; Dong, Y.; Jelinek, J.; Cesaroni, M.; Issa, J.-P.; et al. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat. Neurosci. 2015, 18, 1746–1755. [Google Scholar] [CrossRef] [Green Version]
- Loh, J.T.; Lim, T.J.F.; Ikumi, K.; Matoba, T.; Janela, B.; Gunawan, M.; Toyama, T.; Bunjamin, M.; Ng, L.G.; Poidinger, M.; et al. Ezh2 Controls Skin Tolerance through Distinct Mechanisms in Different Subsets of Skin Dendritic Cells. iScience 2018, 10, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874, Corrigendum in 2007, 446, 824. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, H.-R.; Taing, K.; Chen, L.; Penney, A. EZH2 Methyltransferase Regulates Neuroinflammation and Neuropathic Pain. Cells 2023, 12, 1058. https://doi.org/10.3390/cells12071058
Weng H-R, Taing K, Chen L, Penney A. EZH2 Methyltransferase Regulates Neuroinflammation and Neuropathic Pain. Cells. 2023; 12(7):1058. https://doi.org/10.3390/cells12071058
Chicago/Turabian StyleWeng, Han-Rong, Kyle Taing, Lawrence Chen, and Angela Penney. 2023. "EZH2 Methyltransferase Regulates Neuroinflammation and Neuropathic Pain" Cells 12, no. 7: 1058. https://doi.org/10.3390/cells12071058