
Microglia Mediated Neuroinflammation in Parkinson’s Disease
 , , , and
, , , and
Abstract
:1. Introduction
2. Neuroinflammation and PD
2.1. Role of Innate and Adaptive Immunity and Their Members
2.2. Role of Inflammatory Mediators
2.3. Role of Neurons
2.4. Role of Neuroinflammation-Triggering Factors
3. Microglia
3.1. Origin and Characteristics of Microglia
3.2. Functions of Microglia
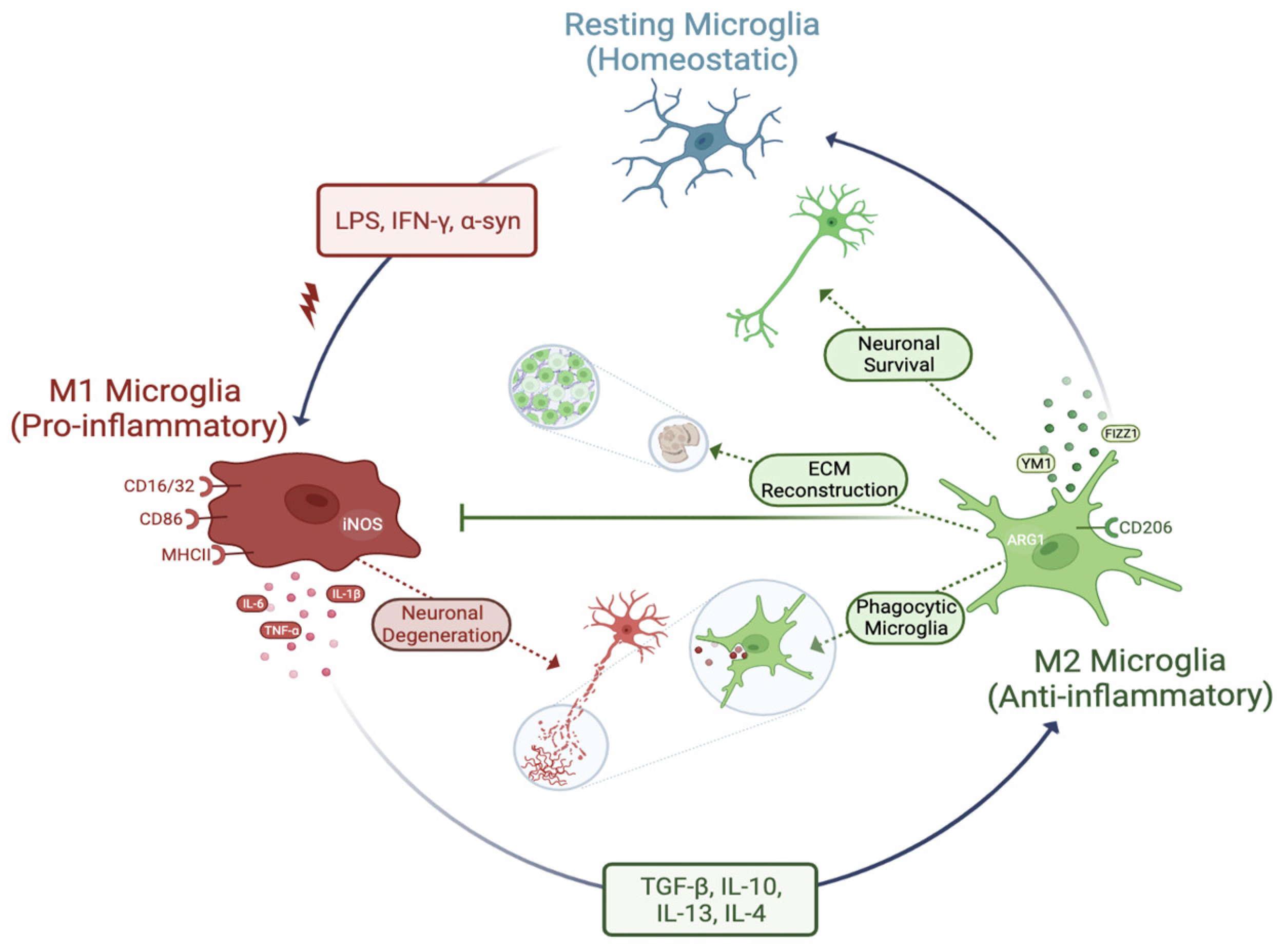
3.3. Phenotypes of Microglia (Activation of Microglia)
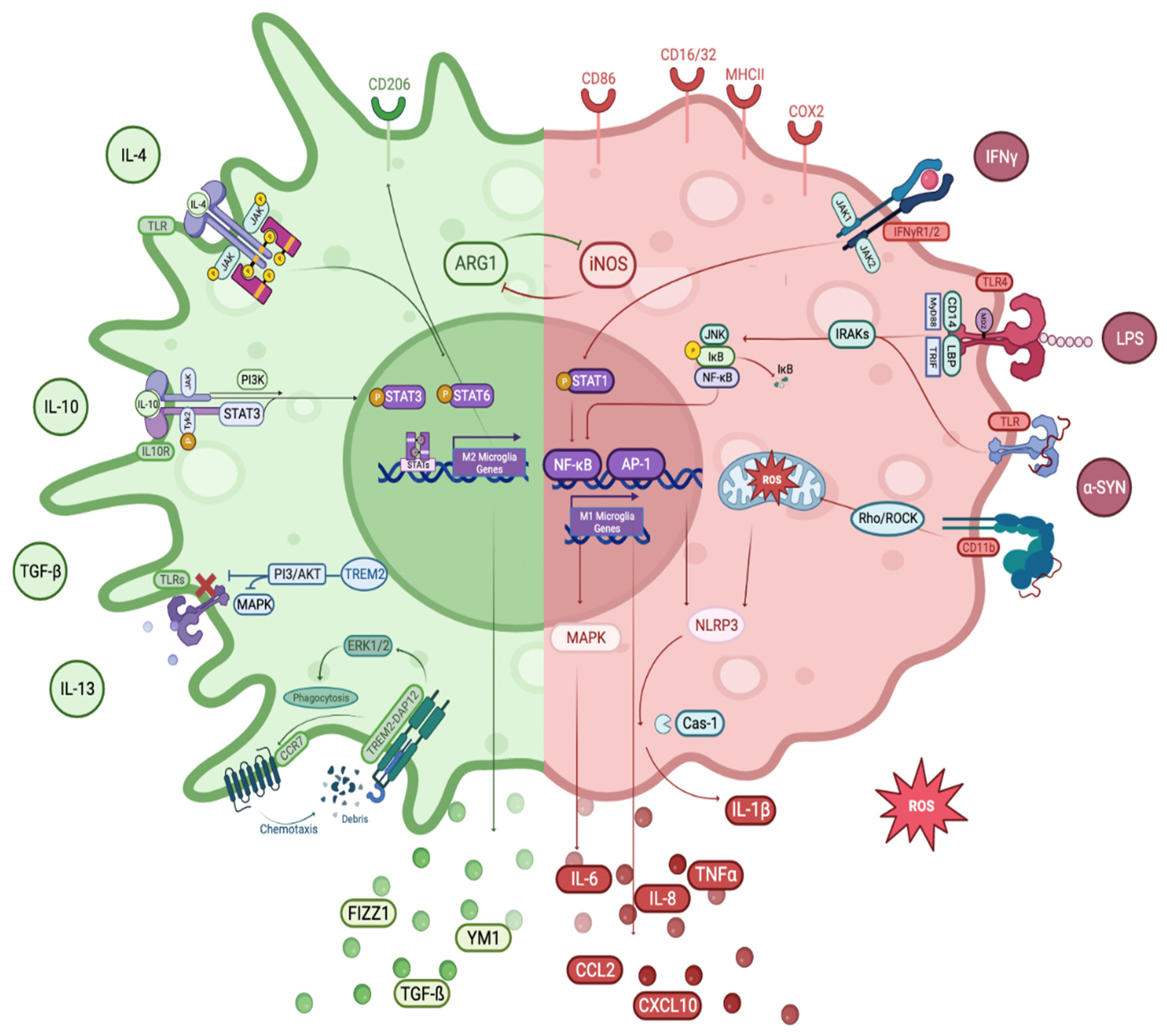
3.3.1. M1 Phenotype
Potent Activators of M1 Phenotype of Microglia:
Toll-like Receptors (TLRs)
TLR4
TLR2
IκB/NF-κB Pathway
MAPK/AP-1 Pathway
3.3.2. M2 Phenotype
3.3.3. M1 to M2 Phenotype Transition
TREM2 Receptors
JAK/STAT Pathway
AMPK-Dependent Pathway
PI3 Kinase/Akt Pathway
Rho/ROCK Pathway
Notch Pathway
4. Evidence of Microglia-Mediated Neuroinflammation in PD
4.1. Human Studies
4.1.1. Postmortem Studies
4.1.2. Patient Studies
Brain PET Imaging Studies
Extracellular Biological Fluid Studies
4.2. Animal Studies
4.2.1. Sporadic Models
4.2.2. Transgenic Models
4.3. In Vitro Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s Disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Wang, W.; Song, N.; Jia, F.; Tang, T.; Bao, W.; Zuo, C.; Xie, J.; Jiang, H. Genomic DNA Levels of Mutant Alpha-synuclein Correlate with Non-Motor Symptoms in an A53T Parkinson’s Disease Mouse Model. Neurochem. Int. 2018, 114, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.J.; Rosier, A.J.; Reich, S.G.; Smith, J.S.; Ehlers, M.D.; Snyder, S.H.; Ravert, H.T.; Dannals, R.F. Positron Emission Tomographic Imaging of the Dopamine Transporter with 11C-WIN 35,428 Reveals Marked Declines in Mild Parkinson’s Disease. Ann. Neurol. 1993, 34, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Todd, K.L.; Lipski, J.; Freestone, P.S. Subthalamic Nucleus Exclusively Evokes Dopamine Release in the Tail of the Striatum. J. Neurochem. 2022, 162, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s Disease: Substantia Nigra Regional Selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The Substantia Nigra of the Human Brain II. Patterns of Loss of Dopamine-Containing Neurons in Parkinson’s Disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Braak, H.; del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Iacono, D.; Geraci-Erck, M.; Rabin, M.L.; Adler, C.H.; Serrano, G.; Beach, T.G.; Kurlan, R. Parkinson Disease and Incidental Lewy Body Disease Just a Question of Time? Neurology 2015, 85, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological Assessment of Parkinson’s Disease: Refining the Diagnostic Criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology Underlying Clinical Variability in Patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- del Rey, N.L.-G.; Quiroga-Varela, A.; Garbayo, E.; Carballo-Carbajal, I.; Fernández-Santiago, R.; Monje, M.H.G.; Trigo-Damas, I.; Blanco-Prieto, M.J.; Blesa, J. Advances in Parkinson’s Disease: 200 Years Later. Front. Neuroanat. 2018, 12, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [Green Version]
- Golbe, L.I.; di Iorio, G.; Bonavita, V.; Miller, D.C.; Duvoisin, R.C. A Large Kindred with Autosomal Dominant Parkinson’s Disease. Ann. Neurol. 1990, 27, 276–282. [Google Scholar] [CrossRef]
- Shprecher, D.R.; Adler, C.H.; Zhang, N.; Hentz, J.G.; Serrano, G.E.; Dugger, B.N.; Shill, H.A.; Savica, R.; Caviness, J.N.; Sabbagh, M.N.; et al. Predicting Alpha-synuclein Pathology by REM Sleep Behavior Disorder Diagnosis. Park. Relat. Disord. 2018, 55, 92–96. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Arotcarena, M.L.; Teil, M.; Dehay, B. Autophagy in synucleinopathy: The Overwhelmed and Defective Machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 Gene Transfer Activates Autophagy and Ameliorates the Neurodegenerative Pathology in α-synuclein Models of Parkinson’s and Lewy Body Diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal. Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons Is Attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic Lysosomal Depletion in Parkinson’s Disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P. Oxidative Mechanisms in Nigral Cell Death in Parkinson’s Disease. Mov. Disord. 1998, 13 (Suppl. S1), 24–34. [Google Scholar]
- Siddiqui, A.; Hanson, I.; Andersen, J.K. Mao-B Elevation Decreases Parkin’s Ability to Efficiently Clear Damaged Mitochondria: Protective Effects of Rapamycin. Free Radic. Res. 2012, 46, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective Autophagy in Parkinson’s Disease: Role of Oxidative Stress. Mol. Neurobiol. 2012, 46, 639–661. [Google Scholar] [CrossRef]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s Disease and Its Potential as Therapeutic Target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef] [PubMed]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.A.; Anthony, D.C. The Acute Inflammatory Response to Intranigral α-synuclein Differs Significantly from Intranigral Lipopolysaccharide and Is Exacerbated by Peripheral Inflammation. J. Neuroinflammation 2011, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, K.; Roy, A.; Banerjee, R.; Choudhury, S.; Mondal, B.; Halder, S.; Basu, P.; Shubham, S.; Dey, S.; Kumar, H. Inflammasome and α-synuclein in Parkinson’s Disease: A Cross-Sectional Study. J. Neuroimmunol. 2020, 338, 577089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulhaq, Z.S.; Garcia, C.P. Inflammation-Related Gene Polymorphisms Associated with Parkinson’s Disease: An Updated Meta-Analysis. Egypt. J. Med. Hum. Genet. 2020, 21, 14. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and Brain, Innate and Adaptive Immunity in Parkinson’s Disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-Faced Microglia: Beneficial and Detrimental Consequences of Microglial Phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Arena, G.; Sharma, K.; Agyeah, G.; Krüger, R.; Grünewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson’s Disease: A Self-Sustained Loop. Curr. Neurol. Neurosci. Rep. 2022, 22, 427–440. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, J. Neuroinflammation in the Central Nervous System: Symphony of Glial Cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes Are Necessary for Blood–Brain Barrier Maintenance in the Adult Mouse Brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The Role of Innate and Adaptive Immunity in Parkinson’s Disease. J. Park. Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef] [Green Version]
- Chitnis, T.; Weiner, H.L. CNS Inflammation and Neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune Cell Regulation of Glia during CNS Injury and Disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef]
- Hua, J.; Yin, N.; Xu, S.; Chen, Q.; Tao, T.; Zhang, J.; Ding, J.; Fan, Y.; Hu, G. Enhancing the Astrocytic Clearance of Extracellular α-synuclein Aggregates by Ginkgolides Attenuates Neural Cell Injury. Cell. Mol. Neurobiol. 2019, 39, 1017–1028. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and Deposition of Extracellular α-synuclein Aggregates in Microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Gruden, M.A.; Casaite, V.; Meskys, R.; Forsgren, L.; Morozova-Roche, L.A. α-synuclein Reactive Antibodies as Diagnostic Biomarkers in Blood Sera of Parkinson’s Disease Patients. PLoS ONE 2011, 6, e18513. [Google Scholar] [CrossRef] [PubMed]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum Interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ Concentrations Are Elevated in Patients with Atypical and Idiopathic Parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Park, J.-Y.; Paik, S.R.; Jou, I.; Park, S.M. Microglial Phagocytosis Is Enhanced by Monomeric α-synuclein, Not Aggregated α-synuclein: Implications for Parkinson’s Disease. Glia 2008, 56, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-Aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-Released Oligomeric α-synuclein Is an Endogenous Agonist of TLR2 for Paracrine Activation of Microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein Activates Microglia: A Process Leading to Disease Progression in Parkinson’s Disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing Alpha synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, K.; Apel, A.; Schulte, C.; Schneiderhan-Marra, N.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.-C.; Cormier, F.; et al. Inflammatory Profile in LRRK2-Associated Prodromal and Clinical PD. J. Neuroinflammation 2016, 13, 122. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.A.; Kannarkat, G.T.; Cintron, A.F.; Butkovich, L.M.; Fraser, K.B.; Chang, J.; Grigoryan, N.; Factor, S.A.; West, A.B.; Boss, J.M.; et al. LRRK2 Levels in Immune Cells Are Increased in Parkinson’s Disease. NPJ Park. Dis. 2017, 3, 11. [Google Scholar] [CrossRef]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much More than Mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Chahine, L.M.; Qiang, J.; Ashbridge, E.; Minger, J.; Yearout, D.; Horn, S.; Colcher, A.; Hurtig, H.I.; Lee, V.M.Y.; van Deerlin, V.M.; et al. Clinical and Biochemical Differences in Patients Having Parkinson Disease with vs without GBA Mutations. JAMA Neurol. 2013, 70, 852–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Nehru, B. Characterization of the Lipopolysaccharide Induced Model of Parkinson’s Disease: Role of Oxidative Stress and Neuroinflammation. Neurochem. Int. 2015, 87, 92–105. [Google Scholar] [CrossRef]
- Berger, J.R.; Nath, A.; Greenberg, R.N.; Andersen, A.H.; Greene, R.A.; Bognar, A.; Avison, M.J. Cerebrovascular Changes in the Basal Ganglia with HIV Dementia. Neurology 2000, 54, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, S.; Zanin, M.; O’Brien, K.; Schultz-Cherry, S.; Smeyne, R.J. Induction of Microglia Activation after Infection with the Non-Neurotropic A/CA/04/2009 H1N1 Influenza Virus. PLoS ONE 2015, 10, e0124047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caggiu, E.; Paulus, K.; Galleri, G.; Arru, G.; Manetti, R.; Sechi, G.P.; Sechi, L.A. Homologous HSV1 and Alpha-synuclein Peptides Stimulate a T Cell Response in Parkinson’s Disease. J. Neuroimmunol. 2017, 310, 26–31. [Google Scholar] [CrossRef]
- Lofrumento, D.D.; Saponaro, C.; Cianciulli, A.; De Nuccio, F.; Mitolo, V.; Nicolardi, G.; Panaro, M.A. MPTP-Induced Neuroinflammation Increases the Expression of pro-Inflammatory Cytokines and Their Receptors in Mouse Brain. Neuroimmunomodulation 2010, 18, 79–88. [Google Scholar] [CrossRef]
- Maitra, U.; Scaglione, M.N.; Chtarbanova, S.; O’Donnell, J.M. Innate Immune Responses to Paraquat Exposure in a Drosophila Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 12714. [Google Scholar] [CrossRef] [Green Version]
- Thakur, P.; Nehru, B. Inhibition of Neuroinflammation and Mitochondrial Dysfunctions by Carbenoxolone in the Rotenone Model of Parkinson’s Disease. Mol. Neurobiol. 2015, 51, 209–219. [Google Scholar] [CrossRef]
- Villarán, R.F.; Espinosa-Oliva, A.M.; Sarmiento, M.; De Pablos, R.M.; Argüelles, S.; Delgado-Cortés, M.J.; Sobrino, V.; Van Rooijen, N.; Venero, J.L.; Herrera, A.J.; et al. Ulcerative Colitis Exacerbates Lipopolysaccharide-Induced Damage to the Nigral Dopaminergic System: Potential Risk Factor in Parkinson’s Disease. J. Neurochem. 2010, 114, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The Gut Microbiota Influences Blood-Brain Barrier Permeability in Mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa Alpha-synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of Resident Microglia in the Normal Adult Mouse Brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Greter, M.; Merad, M. Regulation of Microglia Development and Homeostasis. Glia 2013, 61, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia Express Distinct M1 and M2 Phenotypic Markers in the Postnatal and Adult Central Nervous System in Male and Female Mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Weiner, H.L. Microglial Signatures and Their Role in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a Unique TGF-β–Dependent Molecular and Functional Signature in Microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M.; el Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a020560. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; el Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Kim, W.R.; Ming, G.L.; Song, H. Activity-Dependent Extrinsic Regulation of Adult Olfactory Bulb and Hippocampal Neurogenesis. Ann. N. Y. Acad. Sci. 2009, 1170, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdinler, P.H.; Macklis, J.D. IGF-I Specifically Enhances Axon Outgrowth of Corticospinal Motor Neurons. Nat. Neurosci. 2006, 9, 1371–1381. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of synapses in Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; McGeer, P.L. Brain Microglia Constitutively Express β-2 Integrins. J. Neuroimmunol. 1990, 30, 81–93. [Google Scholar] [CrossRef]
- Pang, Y.; Fan, L.W.; Tien, L.T.; Dai, X.; Zheng, B.; Cai, Z.; Lin, R.C.S.; Bhatt, A. Differential Roles of Astrocyte and Microglia in Supporting Oligodendrocyte Development and Myelination in Vitro. Brain Behav. 2013, 3, 503–514. [Google Scholar] [CrossRef]
- Djannatian, M.; Weikert, U.; Safaiyan, S.; Wrede, C.; Deichsel, C.; Kislinger, G.; Ruhwedel, T.; Campbell, D.S.; van Ham, T.; Schmid, B.; et al. Myelin Biogenesis Is Associated with Pathological Ultrastructure That Is Resolved by Microglia during Development. bioRxiv 2021. [Google Scholar] [CrossRef]
- Salvi, V.; Sozio, F.; Sozzani, S.; del Prete, A. Role of Atypical Chemokine Receptors in Microglial Activation and Polarization. Front. Aging Neurosci. 2017, 9, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial Phenotypes in Parkinson’s Disease and Animal Models of the Disease. Prog. Neurobiol. 2017, 155, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.S. Microglia in Parkinson’s Disease. In Neuroglia in Neurodegenerative Diseases; Verkhratsky, A., Ho, M.S., Zorec, R., Parpura, V., Eds.; Springer: Singapore, 2019; pp. 335–353. ISBN 9789811399138. [Google Scholar]
- Loane, D.J.; Byrnes, K.R. Role of Microglia in Neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chiba, K. Diversity and Plasticity of Microglial Cells in Psychiatric and Neurological Disorders. Pharmacol. Ther. 2015, 154, 21–35. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Franco, R.; Fernández-Suárez, D. Alternatively Activated Microglia and Macrophages in the Central Nervous System. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κB by Toll-like Receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial α-synuclein/TLRs/NF-KappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α-synuclein, an Inflammatory Response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.R.; Kang, S.J.; Kim, J.M.; Lee, S.J.; Jou, I.; Joe, E.H.; Park, S.M. FcγRIIB Mediates the Inhibitory Effect of Aggregated α-synuclein on Microglial Phagocytosis. Neurobiol. Dis. 2015, 83, 90–99. [Google Scholar] [CrossRef]
- Li, R.; Huang, Y.G.; Fang, D.; Le, W.D. (−)-Epigallocatechin Gallate Inhibits Lipopolysaccharide-Induced Microglial Activation and Protects against Inflammation-Mediated Dopaminergic Neuronal Injury. J. Neurosci. Res. 2004, 78, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR Signaling Pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ivashkiv, L.B. Cross-Regulation of Signaling Pathways by Interferon-γ: Implications for Immune Responses and Autoimmune Diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining GM-CSF– and Macrophage-CSF– Dependent Macrophage Responses by In Vitro Models. J. Immunol. 2012, 188, 5752–5765. [Google Scholar] [CrossRef] [Green Version]
- Weisser, S.B.; McLarren, K.W.; Kuroda, E.; Sly, L.M. Generation and Characterization of Murine Alternatively Activated Macrophages. In Basic Cell Culture Protocols; Helgason, C.D., Miller, C.L., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 225–239. ISBN 978-1-62703-128-8. [Google Scholar]
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs1. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, M.; Triantafilou, K. Heat-Shock Protein 70 and Heat-Shock Protein 90 Associate with Toll-like Receptor 4 in Response to Bacterial Lipopolysaccharide. Biochem. Soc. Trans. 2004, 32, 636–639. [Google Scholar] [CrossRef] [Green Version]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan Activate Dendritic Cells via Toll-like Receptor 4; Rockefeller University Press: New York, NY, USA, 2002; Volume 195. [Google Scholar]
- Zhao, X.D.; Wang, F.X.; Cao, W.F.; Zhang, Y.H.; Li, Y. TLR4 Signaling Mediates AP-1 Activation in an MPTP-Induced Mouse Model of Parkinson’s Disease. Int. Immunopharmacol. 2016, 32, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Jana, M.; Majumder, M.; Mondal, S.; Roy, A.; Pahan, K. Selective Targeting of the TLR2/MyD88/NF-ΚB Pathway Reduces α-synuclein Spreading in Vitro and in Vivo. Nat. Commun. 2021, 12, 5382. [Google Scholar] [CrossRef]
- Baldwin, A.S. THE NF-ΚB AND IκB PROTEINS: New Discoveries and Insights. Annu. Rev. Immunol. 1996, 14, 649–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic Programming of Systemic Ageing Involving IKK-β, NF-ΚB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible New Role for NF-ΚB in the Resolution of Inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. IκB Kinases: Key Regulators of the NF-ΚB Pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. “Good” and “Bad” Microglia in Parkinson′s Disease: An Understanding of Homeostatic Mechanisms in Immunomodulation. In Inflammation in Parkinson’s Disease: Scientific and Clinical Aspects; Thomas, M., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 105–126. ISBN 9783319080468. [Google Scholar]
- Yan, A.; Zhang, Y.; Lin, J.; Song, L.; Wang, X.; Liu, Z. Partial Depletion of Peripheral M1 Macrophages Reverses Motor Deficits in MPTP-Treated Mouse by Suppressing Neuroinflammation and Dopaminergic Neurodegeneration. Front. Aging Neurosci. 2018, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Compromised MAPK Signaling in Human Diseases: An Update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK Cascades: Signaling Components, Nuclear Roles and Mechanisms of Nuclear Translocation. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.-K. c-Jun N-Terminal Kinases (JNKs) Mediate pro-Inflammatory Actions of Microglia. Glia 2005, 50, 235–246. [Google Scholar] [CrossRef]
- Xing, B.; Bachstetter, A.D.; van Eldik, L.J. Microglial P38α MAPK Is Critical for LPS-Induced Neuron Degeneration, through a Mechanism Involving TNFα. Mol. Neurodegener. 2011, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-N.; Kim, S.-U.; Lee, M.-S.; Kim, S.-K.; Kim, J.-M.; Yim, M.; Yu, D.-Y.; Lee, D.-S. Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase-Dependent Activation of Phosphoinositide 3-Kinase and P38 Mitogen-Activated Protein Kinase Signal Pathways Is Required for Lipopolysaccharide-Induced Microglial Phagocytosis. Biol. Pharm. Bull. 2008, 31, 1711–1715. [Google Scholar] [CrossRef] [Green Version]
- Dheen, S.T.; Kaur, C.; Ling, E.-A. Microglial Activation and Its Implications in the Brain Diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell. Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colton, C.A. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, E.D.; Maresz, K.; Tan, Y.; Dittel, B.N. CNS-Derived Interleukin-4 Is Essential for the Regulation of Autoimmune Inflammation and Induces a State of Alternative Activation in Microglial Cells. J. Neurosci. 2007, 27, 10714–10721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raes, G.; Noël, W.; Beschin, A.; Brys, L.; de Baetselier, P.; Hassanzadeh, G.G. FIZZ1 and Ym as Tools to Discriminate between Differentially Activated Macrophages. Dev. Immunol. 2002, 9, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States. Br. J. Pharm. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Li, J.; Wu, H.; Peng, Y.; Fan, L.; Chen, J.; Gu, C.; Yan, F.; Wang, L.; et al. The Polarization States of Microglia in TBI: A New Paradigm for Pharmacological Intervention. Neural Plast. 2017, 2017, 5405104. [Google Scholar] [CrossRef] [Green Version]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of Microglia in Brain Development, Tissue Maintenance and Repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [Green Version]
- Piccio, L.; Buonsanti, C.; Mariani, M.; Cella, M.; Gilfillan, S.; Cross, A.H.; Colonna, M.; Panina-Bordignon, P. Blockade of TREM-2 Exacerbates Experimental Autoimmune Encephalomyelitis. Eur. J. Immunol. 2007, 37, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A Dap12-Mediated Pathway Regulates Expression of Cc Chemokine Receptor 7 and Maturation of Human Dendritic Cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting Edge: TREM-2 Attenuates Macrophage Activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Wang, X.; Hahn, W.C. Genomic Insights into WNT/β-Catenin Signaling. Trends Pharm. Sci. 2014, 35, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Yang, X.; Xu, S.; Qian, Y.; Xiao, Q. Resveratrol Regulates Microglia M1/M2 Polarization via PGC-1α in Conditions of Neuroinflammatory Injury. Brain Behav. Immun. 2017, 64, 162–172. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Varinou, L.; Ramsauer, K.; Karaghiosoff, M.; Kolbe, T.; Pfeffer, K.; Müller, M.; Decker, T. Phosphorylation of the Stat1 Transactivation Domain Is Required for Full-Fledged IFN-γ-Dependent Innate Immunity. Immunity 2003, 19, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional Regulation of Macrophage Polarization: Enabling Diversity with Identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Zhang, W.; Cao, Q.; Zou, L.; Fan, X.; Qi, C.; Yan, Y.; Song, B.; Wu, B. JAK2/STAT3 Pathway Regulates Microglia Polarization Involved in Hippocampal Inflammatory Damage Due to Acute Paraquat Exposure. Ecotoxicol. Env. Saf. 2022, 234, 113372. [Google Scholar] [CrossRef] [PubMed]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The Role of JAK/STAT Signaling Pathway and Its Inhibitors in Diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef]
- Chen, H.; O’Reilly, E.J.; Schwarzschild, M.A.; Ascherio, A. Peripheral Inflammatory Biomarkers and Risk of Parkinson’s Disease. Am. J. Epidemiol. 2008, 167, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of Interferon-γ in Microglial-Mediated Loss of Dopaminergic Neurons. J. Neurosci. 2007, 27, 3328. [Google Scholar] [CrossRef] [Green Version]
- Tran, Q.-K.; Ohashi, K.; Watanabe, H. Calcium Signalling in Endothelial Cells. Cardiovasc. Res. 2000, 48, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca2+/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef] [Green Version]
- Trefts, E.; Shaw, R.J. AMPK: Restoring Metabolic Homeostasis over Space and Time. Mol. Cell. 2021, 81, 3677–3690. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Xu, Y.; Ruan, W.; Wang, H.; Zhang, Y.; Saavedra, J.M.; Zhang, L.; Huang, Z.; Pang, T. A Dual AMPK/Nrf2 Activator Reduces Brain Inflammation After Stroke by Enhancing Microglia M2 Polarization. Antioxid. Redox Signal. 2017, 28, 141–163. [Google Scholar] [CrossRef]
- Tsai, C.-F.; Chen, G.-W.; Chen, Y.-C.; Shen, C.-K.; Lu, D.-Y.; Yang, L.-Y.; Chen, J.-H.; Yeh, W.-L. Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients 2022, 14, 67. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease. J. Park. Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-J.; Chern, Y. AMPK-Mediated Regulation of Neuronal Metabolism and Function in Brain Diseases. J. Neurogenet. 2015, 29, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Qi, B.; Xiaoxiang, W.; Xu, J.; Liu, X. Baicalein Increases Cisplatin Sensitivity of A549 Lung Adenocarcinoma Cells via PI3K/Akt/NF-ΚB Pathway. Biomed. Pharmacother. 2017, 90, 677–685. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, X.; Shaikh, A.S.; Zang, J.; Xu, W.; Zhang, Y. JAK/STAT Signal Transduction: Promising Attractive Targets for Immune, Inflammatory and Hematopoietic Diseases. Curr. Drug. Targets 2017, 19, 487–500. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules 2020, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Linton, M.F.; Moslehi, J.J.; Babaev, V.R. Akt Signaling in Macrophage Polarization, Survival, and Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 2703. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.A.; Henry, R.J.; Blanchard, A.C.; Stoica, B.A.; Loane, D.J.; Faden, A.I. Enhanced Akt/GSK-3β/CREB Signaling Mediates the Anti-Inflammatory Actions of MGluR5 Positive Allosteric Modulators in Microglia and Following Traumatic Brain Injury in Male Mice. J. Neurochem. 2021, 156, 225–248. [Google Scholar] [CrossRef]
- Hoogland, I.C.M.; Westhoff, D.; Engelen-Lee, J.-Y.; Melief, J.; Valls Serón, M.; Houben-Weerts, J.H.M.P.; Huitinga, I.; van Westerloo, D.J.; van der Poll, T.; van Gool, W.A.; et al. Microglial Activation After Systemic Stimulation with Lipopolysaccharide and Escherichia Coli. Front. Cell. Neurosci. 2018, 12, 110. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-Kinase/ROCK: A Key Regulator of the Cytoskeleton and Cell Polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Stankiewicz, T.R.; Linseman, D.A. Rho Family GTPases: Key Players in Neuronal Development, Neuronal Survival, and Neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Song, S.; Zhang, Y.; Ge, Y.; Fang, X.; Huang, T.; Du, J.; Gao, J. Inhibition of the Rho/Rho Kinase Pathway Prevents Lipopolysaccharide-Induced Hyperalgesia and the Release of TNF-α and IL-1β in the Mouse Spinal Cord. Sci. Rep. 2015, 5, 14553. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sun, Z.; Jin, M.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. Inhibition of AGEs/RAGE/Rho/ROCK Pathway Suppresses Non-Specific Neuroinflammation by Regulating BV2 Microglial M1/M2 Polarization through the NF-ΚB Pathway. J. Neuroimmunol. 2017, 305, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.-S.; Park, S.C. Chemical Screening Identifies ROCK as a Target for Recovering Mitochondrial Function in Hutchinson-Gilford Progeria syndrome. Aging Cell. 2017, 16, 541–550. [Google Scholar] [CrossRef]
- Roser, A.-E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsell, C.E.; Boulter, J.; DiSibio, G.; Gossler, A.; Weinmaster, G. Expression Patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 Genes Identify Ligand–Receptor Pairs That May Function in Neural Development. Mol. Cell. Neurosci. 1996, 8, 14–27. [Google Scholar] [CrossRef]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 Signalling Requires Ligand-Induced Proteolytic Release of Intracellular Domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Kovall, R.A. More Complicated than It Looks: Assembly of Notch Pathway Transcription Complexes. Oncogene 2008, 27, 5099–5109. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in Oncology: The Path Forward. Nat. Rev. Drug. Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Wu, F.; Luo, T.; Mei, Y.; Liu, H.; Dong, J.; Fang, Y.; Peng, J.; Guo, Y. Simvastatin Alters M1/M2 Polarization of Murine BV2 Microglia via Notch Signaling. J. Neuroimmunol. 2018, 316, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.; Zhang, S.-L.; Yuan, X.; Mo, W.-Q.; Wei, F.; Zhao, S.-N.; Yang, W.; Liu, H.; Rong, X. Liraglutide Lowers Body Weight Set Point in DIO Rats and Its Relationship with Hypothalamic Microglia Activation. Obesity 2020, 28, 122–131. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of Major Histocompatibility Complex Class II-Positive Microglia and Cytokine Profile of Parkinson’s Disease Brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial Inflammation in the Parkinsonian Substantia Nigra: Relationship to Alpha-synuclein Deposition. J. Neuroinflammation 2005, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.D.; Glanzer, J.G.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.P.; Mosley, R.L.; et al. Nitrated Alpha-synuclein-Activated Microglial Profiling for Parkinson’s Disease. J. Neurochem. 2008, 104, 1504–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smajić, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-Cell Sequencing of Human Midbrain Reveals Glial Activation and a Parkinson-Specific Neuronal State. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell. 2019, 179, 1609–1622.e16. [Google Scholar] [CrossRef] [Green Version]
- Dupont, A.-C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Tronel, C.; Arlicot, N. Translocator Protein-18 KDa (TSPO) Positron Emission Tomography (PET) Imaging and Its Clinical Impact in Neurodegenerative Diseases. Int. J. Mol. Sci. 2017, 18, 785. [Google Scholar] [CrossRef] [Green Version]
- Braestrup, C.; Albrechtsen, R.; Squires, R.F. High Densities of Benzodiazepine Receptors in Human Cortical Areas. Nature 1977, 269, 702–704. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C.; Tonietto, M.; Bottlaender, M.; Gervais, P.; Kuhnast, B.; Peyronneau, M.A.; Barret, O.; Lagarde, J.; et al. Increased Microglial Activation in Patients with Parkinson Disease Using [18F]-DPA714 TSPO PET Imaging. Park. Relat. Disord. 2021, 82, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Qiao, H.-W.; Song, T.-B.; Liu, X.-L.; Yao, Y.-X.; Zhao, C.-S.; Barret, O.; Xu, S.-L.; Cai, Y.-N.; Tamagnan, G.D.; et al. Brain Microglia Activation and Peripheral Adaptive Immunity in Parkinson’s Disease: A Multimodal PET Study. J. Neuroinflammation 2022, 19, 209. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Cheon, S.-M.; Bae, H.-R.; Kim, S.-H.; Kim, J.W. Elevated Levels of α-synuclein Oligomer in the Cerebrospinal Fluid of Drug-Naïve Patients with Parkinson’s Disease. J. Clin. Neurol. 2011, 7, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Waragai, M.; Wei, J.; Fujita, M.; Nakai, M.; Ho, G.J.; Masliah, E.; Akatsu, H.; Yamada, T.; Hashimoto, M. Increased Level of DJ-1 in the Cerebrospinal Fluids of Sporadic Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2006, 345, 967–972. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum Immune Markers and Disease Progression in an Incident Parkinson’s Disease Cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Bruggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial Damage-Associated Inflammation Highlights Biomarkers in PRKN/PINK1 Parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Kohutnicka, M.; Kurkowska-Jastrzȩbska, I.; Członkowski, A. Microglial Reaction in MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Induced Parkinson’s Disease Mice Model. Neurodegeneration 1996, 5, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Smeyne, R.J.; Jackson-Lewis, V. The MPTP Model of Parkinson’s Disease. Mol. Brain Res. 2005, 134, 57–66. [Google Scholar] [CrossRef]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice Deficient in TNF Receptors Are Protected against Dopaminergic Neurotoxicity: Implications for Parkinson’s Disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [Green Version]
- Kurkowska-Jastrzebska, I.; Wrońska, A.; Kohutnicka, M.S.; Czlonkowski, A.; Czlonkowska, A. The Inflammatory Reaction Following 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Intoxication in Mouse. Exp. Neurol. 1999, 156, 50–61. [Google Scholar] [CrossRef]
- Cicchetti, F.; Brownell, A.L.; Williams, K.; Chen, Y.I.; Livni, E.; Isacson, O. Neuroinflammation of the Nigrostriatal Pathway during Progressive 6-OHDA Dopamine Degeneration in Rats Monitored by Immunohistochemistry and PET Imaging. Eur. J. Neurosci. 2002, 15, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Relationship between Microglial Activation and Dopaminergic Neuronal Loss in the Substantia Nigra: A Time Course Study in a 6-Hydroxydopamine Model of Parkinson’s Disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide Intranigral Injection Induces Inflammatory Reaction and Damage in Nigrostriatal Dopaminergic System. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.S.; Liu, B. Microglial Activation-Mediated Delayed and Progressive Degeneration of Rat Nigral Dopaminergic Neurons: Relevance to Parkinson’s Disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-G.; Mohney, R.P.; Wilson, B.; Jeohn, G.-H.; Liu, B.; Hong, J.-S. Regional Difference in Susceptibility to Lipopolysaccharide-Induced Neurotoxicity in the Rat Brain: Role of Microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Uriarte Huarte, O.; Kyriakis, D.; Heurtaux, T.; Pires-Afonso, Y.; Grzyb, K.; Halder, R.; Buttini, M.; Skupin, A.; Mittelbronn, M.; Michelucci, A. Single-Cell Transcriptomics and In Situ Morphological Analyses Reveal Microglia Heterogeneity Across the Nigrostriatal Pathway. Front. Immunol. 2021, 12, 639613. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia Affect α-synuclein Cell-to-Cell Transfer in a Mouse Model of Parkinson’s Disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef]
- He, Q.; Li, Y.; Guo, S.; Wang, Y.; Lin, W.; Zhang, Q.; Wang, J.; Ma, C.; Xiao, B.-G. Inhibition of Rho-Kinase by Fasudil Protects Dopamine Neurons and Attenuates Inflammatory Response in an Intranasal Lipopolysaccharide-Mediated Parkinson’s Model. Eur. J. Neurosci. 2016, 43, 41–52. [Google Scholar] [CrossRef]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; López-Royo, T.; Casarejos, M.; Lanciego, J.L.; Lastres-Becker, I. Cx3cr1-Deficiency Exacerbates Alpha-synuclein-A53T Induced Neuroinflammation and Neurodegeneration in a Mouse Model of Parkinson’s Disease. Glia 2018, 66, 1752–1762. [Google Scholar] [CrossRef]
- Chesselet, M.-F.; Richter, F.; Zhu, C.; Magen, I.; Watson, M.B.; Subramaniam, S.R. A Progressive Mouse Model of Parkinson’s Disease: The Thy1-Asyn (“Line 61”) Mice. Neurotherapeutics 2012, 9, 297–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-Specific Microglial Activation in Young Mice over-Expressing Human Wildtype Alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human α-synuclein-Harboring Familial Parkinson’s Disease-Linked Ala-53 → Thr Mutation Causes Neurodegenerative Disease with α-synuclein Aggregation in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.; Casaceli, C.; Colla, E.; Lee, M.K.; Palaniappan, C.; Federoff, H.J. Wild-Type and Mutant α-synuclein Induce a Multi-Component Gene Expression Profile Consistent with Shared Pathophysiology in Different Transgenic Mouse Models of PD. Exp. Neurol. 2007, 204, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. synuclein Activates Microglia in a Model of Parkinson’s Disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Tofaris, G.K.; Garcia Reitböck, P.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological Changes in Dopaminergic Nerve Cells of the Substantia Nigra and Olfactory Bulb in Mice Transgenic for Truncated Human α-synuclein(1–120): Implications for Lewy Body Disorders. J. Neurosci. 2006, 26, 3942–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmer, K.L.; Waxman, E.A.; Covy, J.P.; Giasson, B.I. E46K Human α-synuclein Transgenic Mice Develop Lewy-like and Tau Pathology Associated with Age-Dependent, Detrimental Motor Impairment. J. Biol. Chem. 2011, 286, 35104–35118. [Google Scholar] [CrossRef] [Green Version]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted Overexpression of Human α-synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-Like Receptor 4 Promotes α-synuclein Clearance and Survival of Nigral Dopaminergic Neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product1. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [CrossRef]
- Solano, R.M.; Casarejos, M.J.; Menéndez-Cuervo, J.; Rodriguez-Navarro, J.A.; García de Yébenes, J.; Mena, M.A. Glial Dysfunction in Parkin Null Mice: Effects of Aging. J. Neurosci. 2008, 28, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Cannon, T.C.; Tran, T.; Ruhn, K.A.; Martinez, T.N.; Hong, J.; Marvin, M.; Hartley, M.; Treviño, I.; OBrien, D.E.; Casey, B.; et al. Parkin Deficiency Increases Vulnerability to Inflammation-Related Nigral Degeneration. J. Neurosci. 2008, 28, 10825–10834. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Navarro, J.A.; Casarejos, M.J.; Menéndez, J.; Solano, R.M.; Rodal, I.; Gómez, A.; de Yébenes, J.G.; Mena, M.A. Mortality, Oxidative Stress and Tau Accumulation during Ageing in Parkin Null Mice. J. Neurochem. 2007, 103, 98–114. [Google Scholar] [CrossRef]
- Daher, J.P.L.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of α-synuclein–Mediated Dopaminergic Neurodegeneration in LRRK2-Deficient Rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Byun, J.-W.; Choi, I.; Kim, B.; Jeong, H.-K.; Jou, I.; Joe, E. PINK1 Deficiency Enhances Inflammatory Cytokine Release from Acutely Prepared Brain Slices. Exp. Neurobiol. 2013, 22, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Chen, C.; Yang, D.; Ding, J.; Wang, G.; Ren, H. DJ-1 Inhibits Microglial Activation and Protects Dopaminergic Neurons in Vitro and in Vivo through Interacting with Microglial P65. Cell. Death Dis. 2021, 12, 715. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.C.; von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C.M. Alpha-synuclein Activates BV2 Microglia Dependent on Its Aggregation State. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef]
- Jin, J.; Shie, F.-S.; Liu, J.; Wang, Y.; Davis, J.; Schantz, A.M.; Montine, K.S.; Montine, T.J.; Zhang, J. Prostaglandin E2 Receptor Subtype 2 (EP2) Regulates Microglial Activation and Associated Neurotoxicity Induced by Aggregated α-synuclein. J. Neuroinflammation 2007, 4, 2. [Google Scholar] [CrossRef]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 Receptor Is Critical in α-synuclein–Mediated Microglial NADPH Oxidase Activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef]
- Ingham, V.; Williams, A.; Bate, C. Glimepiride Reduces CD14 Expression and Cytokine Secretion from Macrophages. J. Neuroinflammation 2014, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chu, C.-H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.-S.; Zhang, J. α-synuclein, a Chemoattractant, Directs Microglial Migration via H2O2-Dependent Lyn Phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Sasaoka, T.; Azuma, S.; Ichikawa, T.; Melrose, H.L.; Farrer, M.J.; Obata, F. Leucine-Rich Repeat Kinase 2 (LRRK2) Regulates α-synuclein Clearance in Microglia. BMC Neurosci. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Yang, M.-S.; Choi, D.; Kim, J.-H.; Kim, H.-S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.-Y.; Joe, E. Impaired Inflammatory Responses in Murine Lrrk2-Knockdown Brain Microglia. PLoS ONE 2012, 7, e34693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.A.; Nguyen, A.D.; Chang, J.; Goldberg, M.S.; Lee, J.-K.; Tansey, M.G. Lipopolysaccharide and Tumor Necrosis Factor Regulate Parkin Expression via Nuclear Factor-Kappa B. PLoS ONE 2011, 6, e23660. [Google Scholar] [CrossRef]
- Trudler, D.; Weinreb, O.; Mandel, S.A.; Youdim, M.B.H.; Frenkel, D. DJ-1 Deficiency Triggers Microglia Sensitivity to Dopamine toward a Pro-Inflammatory Phenotype That Is Attenuated by Rasagiline. J. Neurochem. 2014, 129, 434–447. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 Deficiency Impairs Autophagy and Reduces Alpha-synuclein Phagocytosis by Microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Büeler, H. Lack of PINK1 Alters Glia Innate Immune Responses and Enhances Inflammation-Induced, Nitric Oxide-Mediated Neuron Death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Study | Participants | Target | Results |
|---|---|---|---|
| Postmortem Studies | |||
| McGeer, P L et al. [75] | 5 PD; 9 DAT; 2 ccerebrovascular accidents; 7 non-neurological controls. | Brain | ↑ Number of Reactive Microglia in SNcp. ↑ MHC-II positive ameboid microglia. |
| Imamura, Kazuhiro et al. [178] | 12 PD; 4 controls. | Brain | ↑ MHC-II positive microglia in SN. ↑ MHC-II positive microglia in hippocampus, cingulate and temporal cortexes. |
| Croisier, Emilie et al. [179] | 37 PD. | Brain | ↓ Dopaminergic neuronal. ↑ α-syn in SN. ↑ CD-86+ phagocytes. |
| Reynolds, Ashley D et al. [180] | 10 PD; 3 AD; 10 controls. | Brain | ↑ NF-κB in SNpc in PD. |
| Smajić, Semra et al. [181] | 5 idiopathic PD; 6 controls. | Brain | ↑ ameboid microglia in SN. |
| Brochard, Vanessa et al. [47] | 14 PD; 8 controls. | Brain | ↑ CD4+ & CD8+ T cells in SN. |
| Brain PET Imaging | |||
| Lavisse, Sonia et al. [185] | 24 PD; 28 controls. | Brain | ↑ reactivated microglia in midbrain. |
| Liu, Shu-Ying et al. [186] | 24 early-stage PD; 23 controls. | Brain | ↑ reactive microglia in putamen. |
| Extracellular Biological fluid studies | |||
| Park, Min Jeong et al. [187] | 23 naïve-drug PD; 28 neurological controls. | CSF | ↑ oligomeric α-syn in PD. |
| Waragai, Masaaki et al. [188] | 40 sporadic PD; 38 controls. | CSF | ↑ DJ-1 in early-stage PD. |
| Williams-Gray, Caroline H et al. [189] | 230 PD; 93 controls. | Serum cytokines | ↑ TNFα, IL-1β, IL-2, and IL-10. |
| Borsche, Max et al. [190] | 15 biallelic PRKN/PINK1 carrier; 19 affected heterozygous PRKN/PINK1 carrier; 15 unaffected heterozygous PRKN/PINK1 carrier; 59 idiopathic PD 90 controls. | Serum IL-6 and cell-free mtDNA | ↑ IL-6 in biallelic PRKN/PINK1 mutations carriers. ↑ cell-free mtDNA in biallelic and heterogeneous PRKN/PINK1 mutation carriers. |
| Liu, Shu-Ying et al. [186] | 24 early-stage PD; 23 controls. | Blood and serum cytokines | ↑ Th1 cells in blood. ↑ IL-10 and IL-17A in serum. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. https://doi.org/10.3390/cells12071012
Isik S, Yeman Kiyak B, Akbayir R, Seyhali R, Arpaci T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells. 2023; 12(7):1012. https://doi.org/10.3390/cells12071012
Chicago/Turabian StyleIsik, Sevim, Bercem Yeman Kiyak, Rumeysa Akbayir, Rama Seyhali, and Tahire Arpaci. 2023. "Microglia Mediated Neuroinflammation in Parkinson’s Disease" Cells 12, no. 7: 1012. https://doi.org/10.3390/cells12071012