
Characterization of BV6-Induced Sensitization to the NK Cell Killing of Pediatric Rhabdomyosarcoma Spheroids

 , , , , and
, , , , and 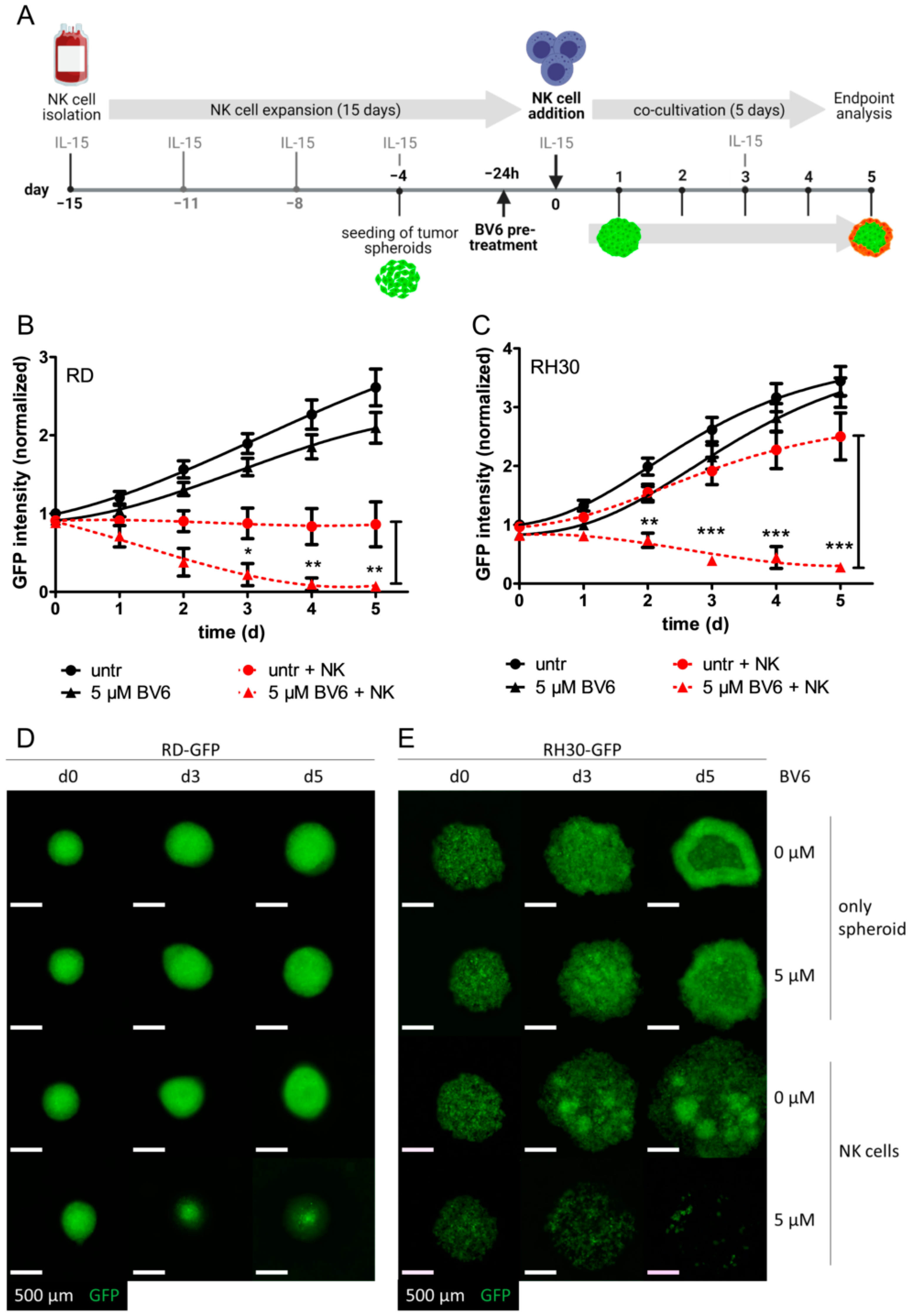{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. NK Cell Enrichment from Peripheral Blood Mononuclear Cells (PBMCs)
2.4. Spheroid Generation and Co-Cultivation
2.5. Immunoblotting
2.6. qRT-PCR
2.7. RNA Sequencing
2.8. siRNA Transfection of RMS-GFP Spheroids
2.9. Statistical Analysis
3. Results
3.1. BV6 Facilitated the Increased NK Cell Killing of RMS-GFP Spheroids
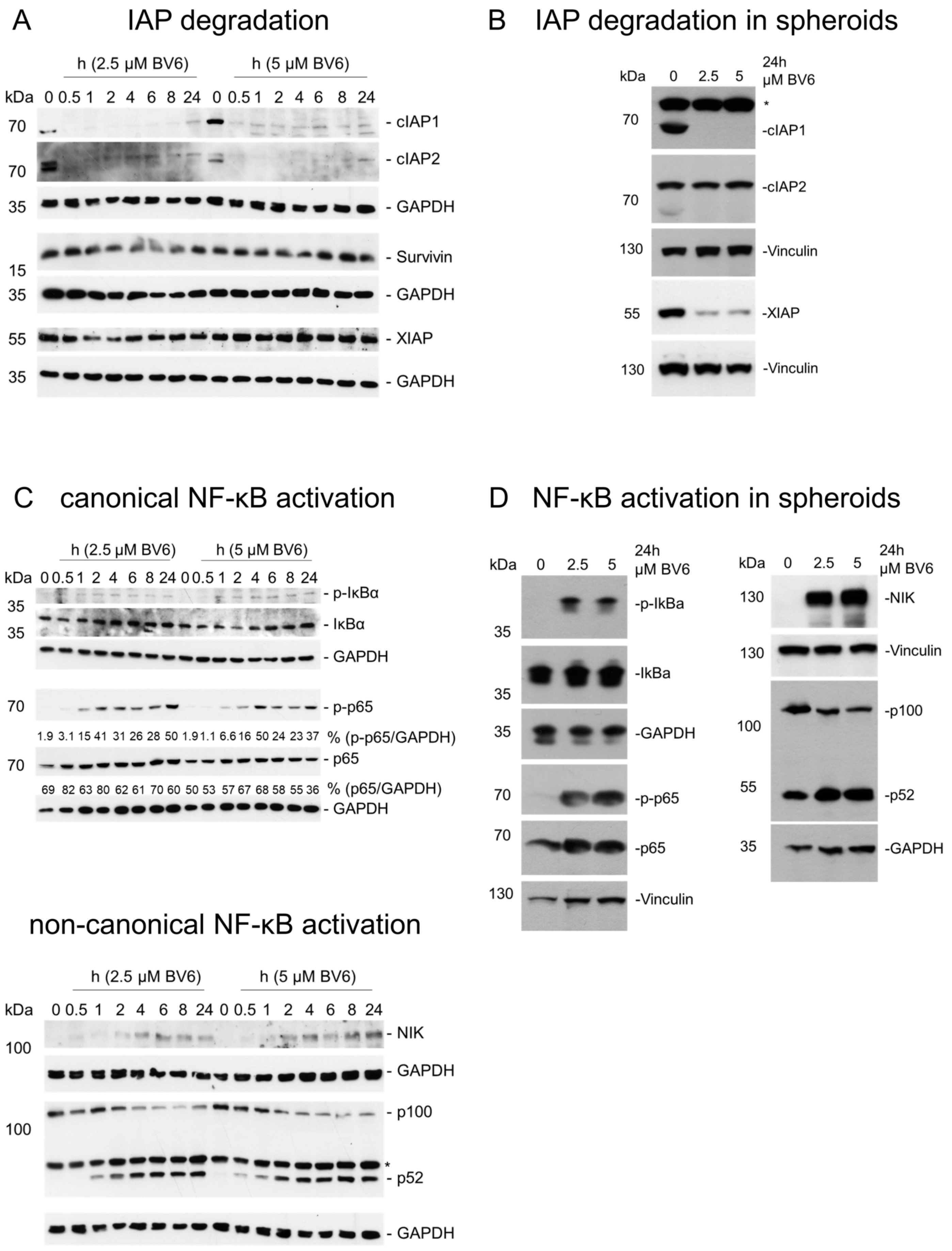
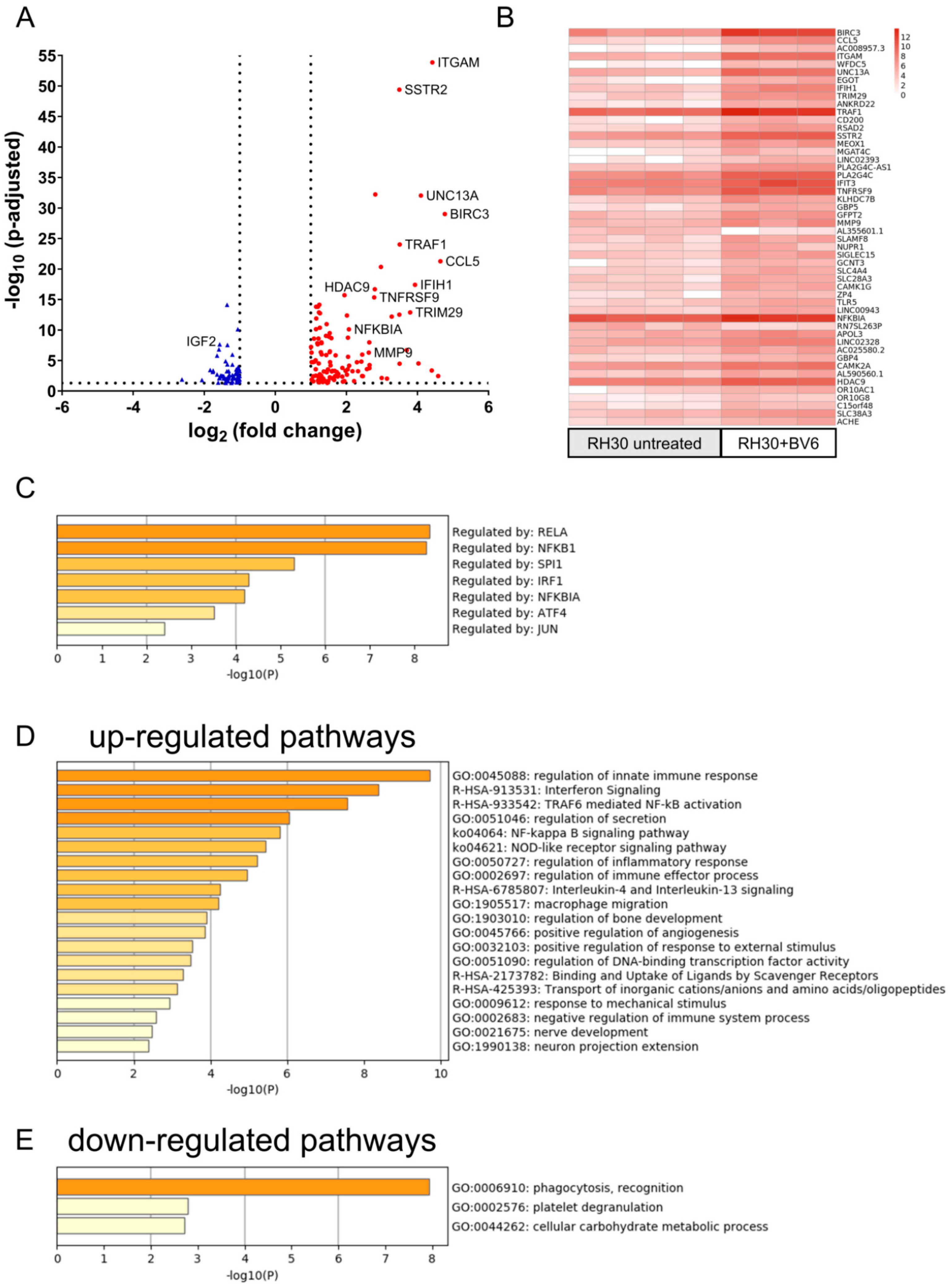
3.2. Activation of NF-κB Signaling Pathways and Transcriptome Regulation by BV6
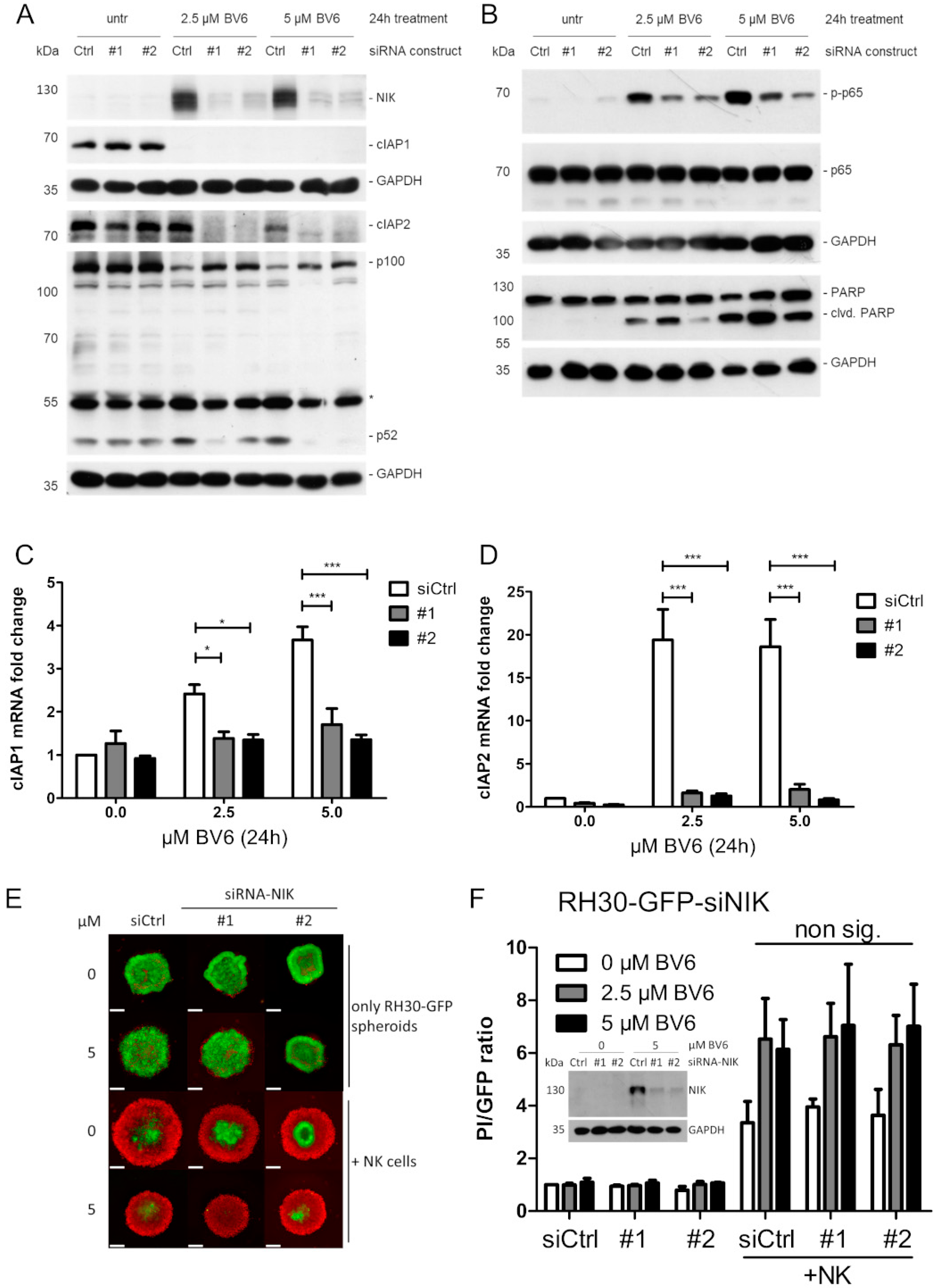
3.3. NIK as Transcriptional Master Regulator of BV6-Induced Transcriptional Change
3.4. Investigation of NK Cell Killing Pathways
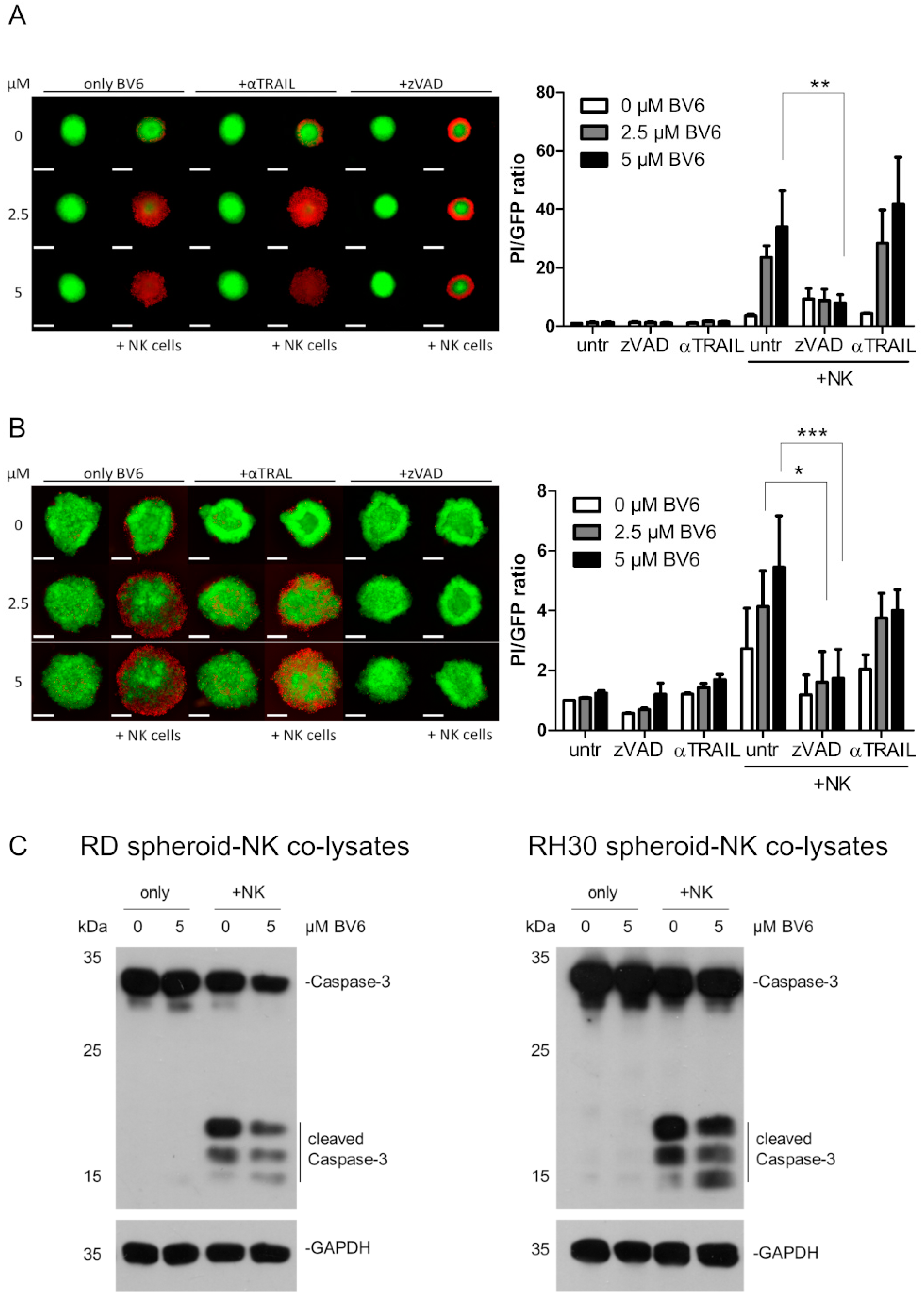
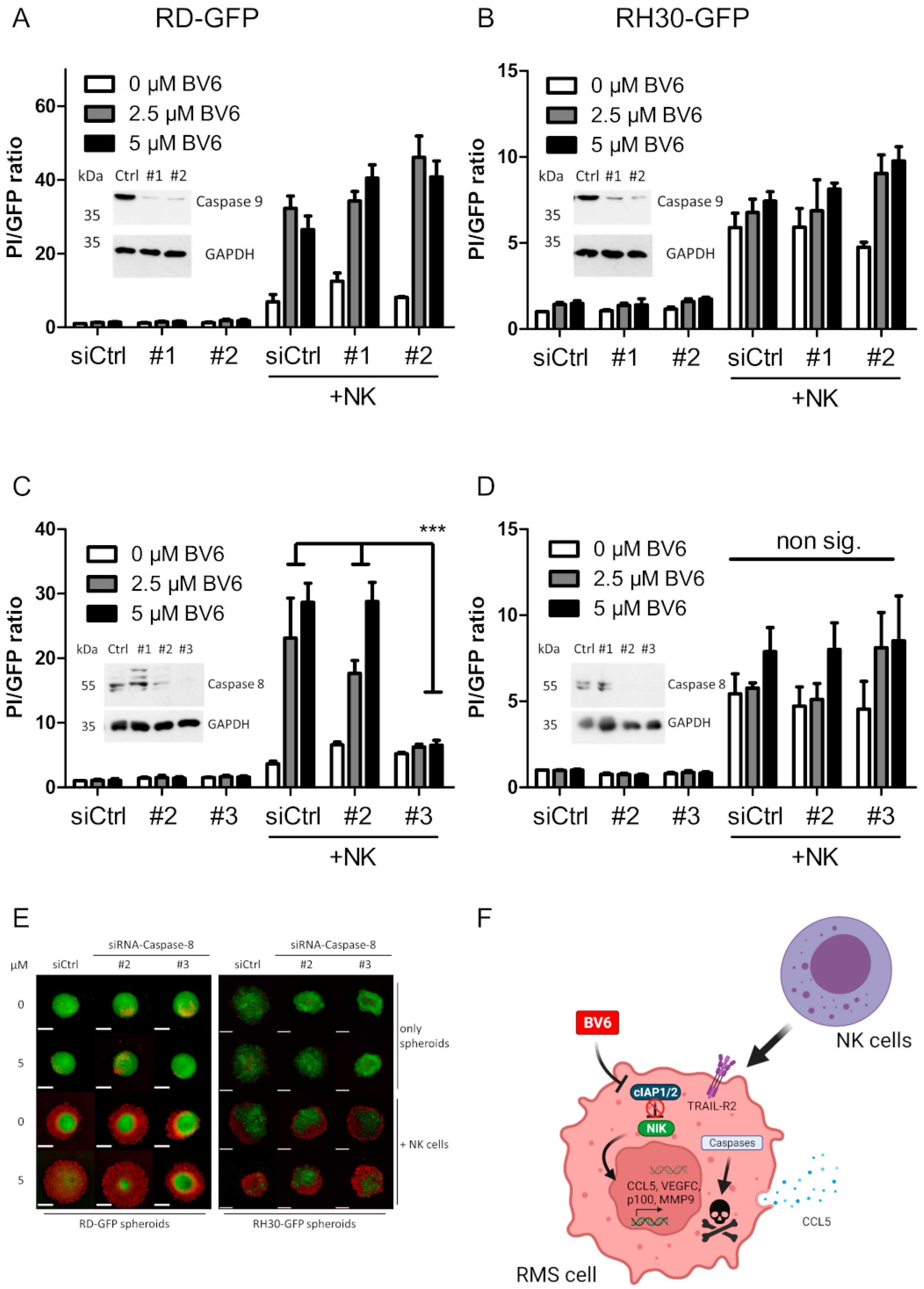
3.5. Dissection of Caspase Involvement in NK Cell-Dependent Killing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leiner, J.; Le Loarer, F. The current landscape of rhabdomyosarcomas: An update. Virchows Arch. 2020, 476, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, I.; Kurzawa, P.; Dopierala, M.; Larque, A.B.; Januszkiewicz-Lewandowska, D. Rhabdomyosarcoma in children—Current pathologic and molecular classification. Pol. J. Pathol. 2018, 69, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.B.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the children’s oncology group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 5. [Google Scholar] [CrossRef]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef]
- Ferrari, A.; Bergamaschi, L.; Chiaravalli, S.; Livellara, V.; Sironi, G.; Nigro, O.; Puma, N.; Gattuso, G.; Morosi, C.; Gasparini, P.; et al. Metastatic rhabdomyosarcoma: Evidence of the impact of radiotherapy on survival. A retrospective single-center experience. Pediatr. Blood Cancer 2022, 69, e29853. [Google Scholar] [CrossRef]
- Hokland, P.; Hokland, M.; Cotter, F. The Nobel Prize for Medicine awarded for cancer therapy by inhibition of negative immune regulation. Br. J. Haematol. 2018, 183, 698–700. [Google Scholar] [CrossRef]
- Chen, L.; Oke, T.; Siegel, N.; Cojocaru, G.; Tam, A.J.; Blosser, R.L.; Swailes, J.; Ligon, J.A.; Lebid, A.; Morris, C.; et al. The Immunosuppressive Niche of Soft-Tissue Sarcomas is Sustained by Tumor-Associated Macrophages and Characterized by Intratumoral Tertiary Lymphoid Structures. Clin. Cancer Res. 2020, 26, 4018–4030. [Google Scholar] [CrossRef]
- Kather, J.N.; Horner, C.; Weis, C.A.; Aung, T.; Vokuhl, C.; Weiss, C.; Scheer, M.; Marx, A.; Simon-Keller, K. CD163+ immune cell infiltrates and presence of CD54+ microvessels are prognostic markers for patients with embryonal rhabdomyosarcoma. Sci. Rep. 2019, 9, 9211. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Ivanova, M.E.; Lukoyanova, N.; Malhotra, S.; Topf, M.; Trapani, J.A.; Voskoboinik, I.; Saibil, H.R. The pore conformation of lymphocyte perforin. Sci. Adv. 2022, 8, eabk3147. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandstrom, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Barbour, V. Celebrating death--the 2002 Nobel prize in physiology or medicine. Lancet 2002, 360, 1117. [Google Scholar] [CrossRef]
- Marx, J.L. The 1988 Nobel Prize for Physiology or Medicine. Science 1988, 242, 516–517. [Google Scholar] [CrossRef]
- Sun, H.; Nikolovska-Coleska, Z.; Lu, J.; Meagher, J.L.; Yang, C.-Y.; Qiu, S.; Tomita, Y.; Ueda, Y.; Jiang, S.; Krajewski, K.; et al. Design, Synthesis, and Characterization of a Potent, Nonpeptide, Cell-Permeable, Bivalent Smac Mimetic That Concurrently Targets Both the BIR2 and BIR3 Domains in XIAP. J. Am. Chem. Soc. 2007, 129, 15279–15294. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Faye, M.D.; Beug, S.T.; Graber, T.E.; Earl, N.; Xiang, X.; Wild, B.; Langlois, S.; Michaud, J.; Cowan, K.N.; Korneluk, R.G.; et al. IGF2BP1 controls cell death and drug resistance in rhabdomyosarcomas by regulating translation of cIAP1. Oncogene 2015, 34, 1532–1541. [Google Scholar] [CrossRef]
- Holt, S.V.; Brookes, K.E.; Dive, C.; Makin, G.W.J. Down-regulation of XIAP by AEG35156 in paediatric tumour cells induces apoptosis and sensitises cells to cytotoxic agents. Oncol. Rep. 2011, 25, 1177–1181. [Google Scholar] [CrossRef]
- Li, J.; Yin, Q.; Wu, H. Structural basis of signal transduction in the TNF receptor superfamily. Adv. Immunol. 2013, 119, 135. [Google Scholar]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef]
- Lee, S.; Challa-Malladi, M.; Bratton, S.B.; Wright, C.W. Nuclear Factor-κB-inducing Kinase (NIK) Contains an Amino-terminal Inhibitor of Apoptosis (IAP)-binding Motif (IBM) That Potentiates NIK Degradation by Cellular IAP1 (c-IAP1). J. Biol. Chem. 2014, 289, 30680–30689. [Google Scholar] [CrossRef] [PubMed]
- Särchen, V.; Shanmugalingam, S.; Kehr, S.; Reindl, L.M.; Greze, V.; Wiedemann, S.; Boedicker, C.; Jacob, M.; Bankov, K.; Becker, N.; et al. Pediatric multicellular tumor spheroid models illustrate a therapeutic potential by combining BH3 mimetics with Natural Killer (NK) cell-based immunotherapy. Cell Death Discov. 2022, 8, 11. [Google Scholar] [CrossRef]
- Fischer, K.; Tognarelli, S.; Roesler, S.; Boedicker, C.; Schubert, R.; Steinle, A.; Klingebiel, T.; Bader, P.; Fulda, S.; Ullrich, E. The Smac Mimetic BV6 Improves NK Cell-Mediated Killing of Rhabdomyosarcoma Cells by Simultaneously Targeting Tumor and Effector Cells. Front. Immunol. 2017, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Rettinger, E.; Glatthaar, A.; Abhari, B.A.; Oelsner, S.; Pfirrmann, V.; Huenecke, S.; Kuci, S.; Kreyenberg, H.; Willasch, A.M.; Klingebiel, T.; et al. SMAC mimetic BV6 enables sensitization of resistant tumor cells but also affects cytokine-induced killer (CIK) cells: A potential challenge for combination therapy. Front. Pediatr. 2014, 2, 75. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Pfannenstiel, V.; Waldmann, A.; Bergs, J.W.J.; Brill, B.; Huenecke, S.; Klingebiel, T.; Rodel, F.; Buchholz, C.J.; Wels, W.S.; et al. A Two-Phase Expansion Protocol Combining Interleukin (IL)-15 and IL-21 Improves Natural Killer Cell Proliferation and Cytotoxicity against Rhabdomyosarcoma. Front. Immunol. 2017, 8, 676. [Google Scholar] [CrossRef]
- Merker, M.; Pfirrmann, V.; Oelsner, S.; Fulda, S.; Klingebiel, T.; Wels, W.S.; Bader, P.; Rettinger, E. Generation and characterization of ErbB2-CAR-engineered cytokine-induced killer cells for the treatment of high-risk soft tissue sarcoma in children. Oncotarget 2017, 8, 66137–66153. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Heinze, A.; Grebe, B.; Bremm, M.; Huenecke, S.; Munir, T.A.; Graafen, L.; Frueh, J.T.; Merker, M.; Rettinger, E.; Soerensen, J.; et al. The Synergistic Use of IL-15 and IL-21 for the Generation of NK Cells From CD3/CD19-Depleted Grafts Improves Their ex vivo Expansion and Cytotoxic Potential against Neuroblastoma: Perspective for Optimized Immunotherapy Post Haploidentical Stem Cell Transplantation. Front. Immunol. 2019, 10, 2816. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Haydn, T.; Schneider, I.; Busch, H.; Boerries, M.; Fulda, S. Smac mimetic induces an early wave of gene expression via NF-kB and AP-1 and a second wave via TNFR1 signaling. Cancer Lett. 2018, 421, 170–185. [Google Scholar] [CrossRef]
- Schmidt, N.; Kowald, L.; van Wijk, S.J.L.; Fulda, S. Differential involvement of TAK1, RIPK1 and NF-kappaB signaling in Smac mimetic-induced cell death in breast cancer cells. Biol. Chem. 2019, 400, 171–180. [Google Scholar] [CrossRef]
- Bhat, H.; Zaun, G.; Hamdan, T.A.; Lang, J.; Adomati, T.; Schmitz, R.; Friedrich, S.K.; Bergerhausen, M.; Cham, L.B.; Li, F.; et al. Arenavirus Induced CCL5 Expression Causes NK Cell-Mediated Melanoma Regression. Front. Immunol. 2020, 11, 1849. [Google Scholar] [CrossRef]
- Maghazachi, A.A.; Al-Aoukaty, A.; Schall, T.J. C-C chemokines induce the chemotaxis of NK and IL-2-activated NK cells. Role for G proteins. J. Immunol. 1994, 153, 4969–4977. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, I.; Roesler, S.; Fulda, S. Identification of DR5 as a critical, NF-kappaB-regulated mediator of Smac-induced apoptosis. Cell Death Dis. 2013, 4, e936. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Belz, K.; Schoeneberger, H.; Wehner, S.; Weigert, A.; Bönig, H.; Klingebiel, T.; Fichtner, I.; Fulda, S. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood 2014, 124, 240–250. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Steigerwald, C.; Rasenberger, B.; Brozovic, A.; Christmann, M. Functional mismatch repair and inactive p53 drive sensitization of colorectal cancer cells to irinotecan via the IAP antagonist BV6. Arch. Toxicol. 2019, 93, 2265–2277. [Google Scholar] [CrossRef]
- Ahmad, I.; Dera, A.; Irfan, S.; Rajagopalan, P.; Ali Beg, M.; Alshahrani, M.; Mir, M.; Abohashrh, M.; Alam, M.; Wahab, S.; et al. BV6 enhances apoptosis in Lung cancer cells by ameliorating caspase expressions through attenuation of XIAP, cIAP-1, and cIAP-2 proteins. J. Can. Res. Ther. 2021, 18, 1651. [Google Scholar] [CrossRef]
- Gallardo-Pérez, J.; Espinosa, M.; Ceballos-Cancino, G.; Daniel, A.; Rodríguez-Enríquez, S.; Aviles, A.; Moreno-Sánchez, R.; Melendez-Zajgla, J.; Maldonado, V. NF-kappa B is required for the development of tumor spheroids. J. Cell. Biochem. 2009, 108, 169–180. [Google Scholar] [CrossRef]
- Opel, D.; Schnaiter, A.; Dodier, D.; Jovanovic, M.; Gerhardinger, A.; Idler, I.; Mertens, D.; Bullinger, L.; Stilgenbauer, S.; Fulda, S. Targeting inhibitor of apoptosis proteins by Smac mimetic elicits cell death in poor prognostic subgroups of chronic lymphocytic leukemia. Int. J. Cancer 2015, 137, 2959–2970. [Google Scholar] [CrossRef]
- Ottaviani, C.; Nasorri, F.; Bedini, C.; de Pita, O.; Girolomoni, G.; Cavani, A. CD56brightCD16(-) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur. J. Immunol. 2006, 36, 118–128. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Liu, H.; Dong, W.; Huang, X.; Yang, D.; Hou, J.; Zhang, X. The SMAC Mimetic APG-1387 Sensitizes Immune-Mediated Cell Apoptosis in Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Pan, R.; Ryan, J.; Pan, D.; Wucherpfennig, K.W.; Letai, A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell 2022, 185, 1521–1538.e18. [Google Scholar] [CrossRef]
- Harada, K.; Toyooka, S.; Shivapurkar, N.; Maitra, A.; Reddy, J.L.; Matta, H.; Miyajima, K.; Timmons, C.F.; Tomlinson, G.E.; Mastrangelo, D.; et al. Deregulation of Caspase 8 and 10 Expression in Pediatric Tumors and Cell Lines1. Cancer Res. 2002, 62, 5897–5901. [Google Scholar]
- Petak, I.; Vernes, R.; Szucs, K.S.; Anozie, M.; Izeradjene, K.; Douglas, L.; Tillman, D.M.; Phillips, D.C.; Houghton, J.A. A caspase-8-independent component in TRAIL/Apo-2L-induced cell death in human rhabdomyosarcoma cells. Cell Death Differ. 2003, 10, 729–739. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Khan, N.; Rickard, J.A.; Etemadi, N.; Lalaoui, N.; Spall, S.K.; Hildebrand, J.M.; Segal, D.; Miasari, M.; Chau, D.; et al. Combination of IAP antagonist and IFNγ activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017, 24, 481–491. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; Macfarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Särchen, V.; Reindl, L.M.; Wiedemann, S.; Shanmugalingam, S.; Bukur, T.; Becker, J.; Suchan, M.; Ullrich, E.; Vogler, M. Characterization of BV6-Induced Sensitization to the NK Cell Killing of Pediatric Rhabdomyosarcoma Spheroids. Cells 2023, 12, 906. https://doi.org/10.3390/cells12060906
Särchen V, Reindl LM, Wiedemann S, Shanmugalingam S, Bukur T, Becker J, Suchan M, Ullrich E, Vogler M. Characterization of BV6-Induced Sensitization to the NK Cell Killing of Pediatric Rhabdomyosarcoma Spheroids. Cells. 2023; 12(6):906. https://doi.org/10.3390/cells12060906
Chicago/Turabian StyleSärchen, Vinzenz, Lisa Marie Reindl, Sara Wiedemann, Senthan Shanmugalingam, Thomas Bukur, Julia Becker, Martin Suchan, Evelyn Ullrich, and Meike Vogler. 2023. "Characterization of BV6-Induced Sensitization to the NK Cell Killing of Pediatric Rhabdomyosarcoma Spheroids" Cells 12, no. 6: 906. https://doi.org/10.3390/cells12060906