
Stemming Tumoral Growth: A Matter of Grotesque Organogenesis

{kind=link}
{kind=link}
{kind=link}
Abstract
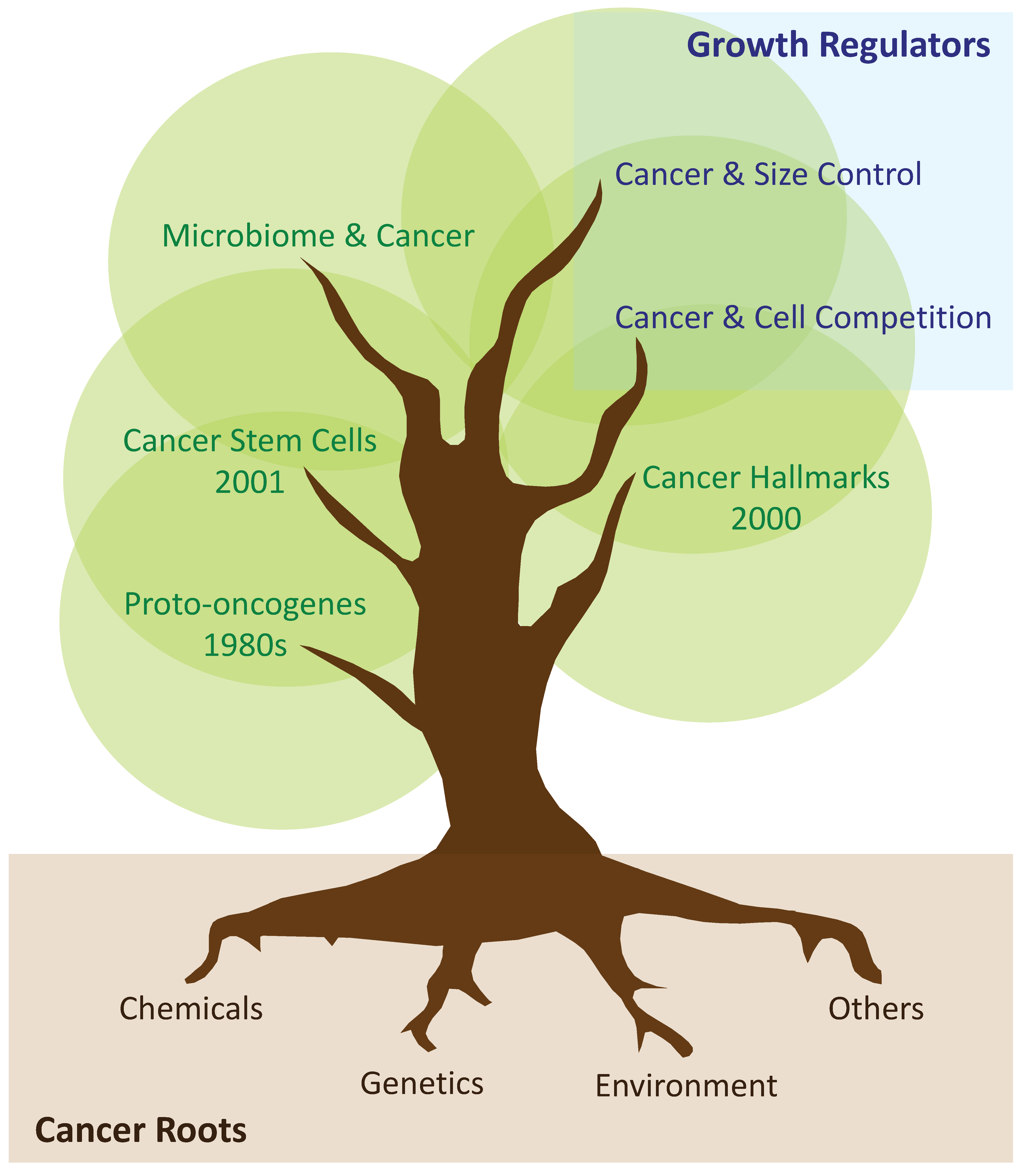
:1. Early Metazoans and Division of Labour
2. The Emergence of Tissue-Specific Stem Cells
3. Tumoral Growth: From Proto-Oncogenes and Hallmarks of Cancer, to the Cancer Stem Cell Hypothesis
4. Tumours as Caricatures of Dysfunctional Organs
5. Cell Competition in Primary and Secondary Cancer Growth
6. Scaling Phenomena during Development, Adulthood, and Tumorigenesis
7. Concluding Remarks and Future Views
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erwin, D.H. Early metazoan life: Divergence, environment and ecology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [Green Version]
- Condie, K.C. Earth as an Evolving Planetary System, 4th ed.; Academic Press: London, UK; San Diego, CA, USA, 2022; p. vii. 397p. [Google Scholar]
- Ruiz-Trillo, I.; Burger, G.; Holland, P.W.; King, N.; Lang, B.F.; Roger, A.J.; Gray, M.W. The origins of multicellularity: A multi-taxon genome initiative. Trends Genet. 2007, 23, 113–118. [Google Scholar] [CrossRef]
- Ros-Rocher, N.; Perez-Posada, A.; Leger, M.M.; Ruiz-Trillo, I. The origin of animals: An ancestral reconstruction of the unicellular-to-multicellular transition. Open Biol. 2021, 11, 200359. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A. The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu. Rev. Genet. 2008, 42, 235–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, T.; King, N. The Origin of Animal Multicellularity and Cell Differentiation. Dev. Cell 2017, 43, 124–140. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.A.; West, S.A. Division of labour and the evolution of extreme specialization. Nat. Ecol. Evol. 2018, 2, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Rueffler, C.; Hermisson, J.; Wagner, G.P. Evolution of functional specialization and division of labor. Proc. Natl. Acad. Sci. USA 2012, 109, E326–E335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, W.E. Origin of Metazoa: Sponges as living fossils. Naturwissenschaften 1998, 85, 11–25. [Google Scholar] [CrossRef]
- Borisenko, I.; Podgornaya, O.I.; Ereskovsky, A.V. From traveler to homebody: Which signaling mechanisms sponge larvae use to become adult sponges? Adv. Protein Chem. Struct. Biol. 2019, 116, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ereskovsky, A.V.; Renard, E.; Borchiellini, C. Cellular and molecular processes leading to embryo formation in sponges: Evidences for high conservation of processes throughout animal evolution. Dev. Genes Evol. 2013, 223, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.D.; Goss, G.G.; Leys, S.P. Freshwater sponges have functional, sealing epithelia with high transepithelial resistance and negative transepithelial potential. PLoS ONE 2010, 5, e15040. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Agoramoorthy, G. Stem cells in the light of evolution. Indian J. Med. Res. 2012, 135, 813–819. [Google Scholar]
- Meyer, S.N.; Amoyel, M.; Bergantinos, C.; de la Cova, C.; Schertel, C.; Basler, K.; Johnston, L.A. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science 2014, 346, 1258236. [Google Scholar] [CrossRef] [Green Version]
- Daley, G.Q. Stem cells and the evolving notion of cellular identity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Breidbach, O. Visions of Nature: The Art and Science of Ernst Haeckel; Prestel: Munich, Germany; London, UK, 2006; 299p. [Google Scholar]
- Ramalho-Santos, M.; Willenbring, H. On the origin of the term “stem cell”. Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, E.A.; Till, J.E. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat Res. 1960, 13, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.J.; Mc, C.E.; Till, J.E. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 1963, 197, 452–454. [Google Scholar] [CrossRef] [Green Version]
- Sharkis, S.J. Canadian stem cell scientists take the prize. Cell 2005, 122, 817–819. [Google Scholar] [CrossRef] [Green Version]
- Funayama, N. The stem cell system in demosponges: Insights into the origin of somatic stem cells. Dev. Growth Differ. 2010, 52, 1–14. [Google Scholar] [CrossRef]
- Muller, W.E. The stem cell concept in sponges (Porifera): Metazoan traits. Semin Cell Dev. Biol. 2006, 17, 481–491. [Google Scholar] [CrossRef]
- Bhoi, S.B.; Kamle, R.A.; Meshram, S.K.; Waghmare, S.A.; Shirsat, K.B. Study of correlation of organ and body weight during autopsy with regard to age and sex in adult population at Solapur region. Indian J. Forensic Community Med. 2017, 4, 115–120. [Google Scholar]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Guerra, E.; Lillo, M.A.; Santamaria, S.; Garcia-Sanz, J.A. Intrinsic cues and hormones control mouse mammary epithelial tree size. FASEB J. 2012, 26, 3844–3853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Trivanovic, D. Adult Stem Cells in Aging. J. Pers. Med. 2022, 12, 795. [Google Scholar] [CrossRef] [PubMed]
- Bhartiya, D. Adult tissue-resident stem cells-fact or fiction? Stem Cell Res. Ther. 2021, 12, 73. [Google Scholar] [CrossRef]
- Kalderon, D. Investigating Adult Stem Cells Through Lineage analyses. Stem Cell Rev. Rep. 2022, 18, 2–22. [Google Scholar] [CrossRef]
- Altshuler, A.; Wickstrom, S.A.; Shalom-Feuerstein, R. Spotlighting adult stem cells: Advances, pitfalls, and challenges. Trends Cell Biol. 2022. [Google Scholar] [CrossRef]
- Pulciani, S.; Santos, E.; Lauver, A.V.; Long, L.K.; Robbins, K.C.; Barbacid, M. Oncogenes in human tumor cell lines: Molecular cloning of a transforming gene from human bladder carcinoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2845–2849. [Google Scholar] [CrossRef] [Green Version]
- Pulciani, S.; Santos, E.; Lauver, A.V.; Long, L.K.; Barbacid, M. Transforming genes in human tumors. J. Cell Biochem. 1982, 20, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Rowan, K. Are cancer stem cells real? After four decades, debate still simmers. J. Natl. Cancer Inst. 2009, 101, 546–547. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.; Ellenbroek, S.I.; Ritsma, L.; Beerling, E.; Vrisekoop, N.; Van Rheenen, J. Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells 2013, 31, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef] [Green Version]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Li, F.; Xu, J.; Liu, S. Cancer Stem Cells and Neovascularization. Cells 2021, 10, 1070. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Investig. 2004, 114, 370–378. [Google Scholar] [CrossRef]
- Skipworth, R.J.; Stewart, G.D.; Dejong, C.H.; Preston, T.; Fearon, K.C. Pathophysiology of cancer cachexia: Much more than host-tumour interaction? Clin. Nutr. 2007, 26, 667–676. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D.; et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waarts, M.R.; Stonestrom, A.J.; Park, Y.C.; Levine, R.L. Targeting mutations in cancer. J. Clin. Investig. 2022, 132, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E. The Concept of “Cancer Stem Cells” in the Context of Classic Carcinogenesis Hypotheses and Experimental Findings. Life 2021, 11, 1308. [Google Scholar] [CrossRef] [PubMed]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef] [PubMed]
- Park, E.M.; Chelvanambi, M.; Bhutiani, N.; Kroemer, G.; Zitvogel, L.; Wargo, J.A. Targeting the gut and tumor microbiota in cancer. Nat. Med. 2022, 28, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mahmood, M.; Reznik, E.; Gammage, P.A. Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer 2022, 8, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wong, L.J.; Mims, M.P. Mitochondrial inheritance and cancer. Transl. Res. 2018, 202, 24–34. [Google Scholar] [CrossRef]
- Dietrich, C.; Weiss, C.; Bockamp, E.; Brisken, C.; Roskams, T.; Morris, R.; Oesch-Bartlomowicz, B.; Oesch, F. Stem cells in chemical carcinogenesis. Arch. Toxicol. 2010, 84, 245–251. [Google Scholar] [CrossRef]
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental risk factors for cancer—Review paper. Ann. Agric. Environ. Med. 2019, 26, 1–7. [Google Scholar] [CrossRef]
- Merino, M.M.; Levayer, R.; Moreno, E. Survival of the Fittest: Essential Roles of Cell Competition in Development, Aging, and Cancer. Trends Cell Biol. 2016, 26, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.; Perez-Garijo, A.; Calleja, M.; Morata, G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 14651–14656. [Google Scholar] [CrossRef] [Green Version]
- Madan, E.; Pelham, C.J.; Nagane, M.; Parker, T.M.; Canas-Marques, R.; Fazio, K.; Shaik, K.; Yuan, Y.; Henriques, V.; Galzerano, A.; et al. Flower isoforms promote competitive growth in cancer. Nature 2019, 572, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Levayer, R.; Hauert, B.; Moreno, E. Cell mixing induced by myc is required for competitive tissue invasion and destruction. Nature 2015, 524, 476–480. [Google Scholar] [CrossRef]
- Gil, J.; Rodriguez, T. Cancer: The Transforming Power of Cell Competition. Curr. Biol. 2016, 26, R164–R166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey-Lozano, N.; La Marca, J.E.; Portela, M.; Richardson, H.E. Drosophila Models of Cell Polarity and Cell Competition in Tumourigenesis. Adv. Exp. Med. Biol. 2019, 1167, 37–64. [Google Scholar] [CrossRef] [PubMed]
- Mumbauer, S.; Pascual, J.; Kolotuev, I.; Hamaratoglu, F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019, 15, e1008396. [Google Scholar] [CrossRef] [Green Version]
- Morata, G.; Ripoll, P. Minutes: Mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 1975, 42, 211–221. [Google Scholar] [CrossRef]
- Morata, G. Cell competition: A historical perspective. Dev. Biol. 2021, 476, 33–40. [Google Scholar] [CrossRef]
- Claveria, C.; Giovinazzo, G.; Sierra, R.; Torres, M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature 2013, 500, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.M.; Rhiner, C.; Lopez-Gay, J.M.; Buechel, D.; Hauert, B.; Moreno, E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell 2015, 160, 461–476. [Google Scholar] [CrossRef] [Green Version]
- Merino, M.M.; Rhiner, C.; Portela, M.; Moreno, E. “Fitness fingerprints” mediate physiological culling of unwanted neurons in Drosophila. Curr. Biol. 2013, 23, 1300–1309. [Google Scholar] [CrossRef] [Green Version]
- Lima, A.; Lubatti, G.; Burgstaller, J.; Hu, D.; Green, A.P.; Di Gregorio, A.; Zawadzki, T.; Pernaute, B.; Mahammadov, E.; Perez-Montero, S.; et al. Cell competition acts as a purifying selection to eliminate cells with mitochondrial defects during early mouse development. Nat. Metab. 2021, 3, 1091–1108. [Google Scholar] [CrossRef]
- Langton, P.F.; Baumgartner, M.E.; Logeay, R.; Piddini, E. Xrp1 and Irbp18 trigger a feed-forward loop of proteotoxic stress to induce the loser status. PLoS Genet. 2021, 17, e1009946. [Google Scholar] [CrossRef]
- Baumgartner, M.E.; Dinan, M.P.; Langton, P.F.; Kucinski, I.; Piddini, E. Proteotoxic stress is a driver of the loser status and cell competition. Nat. Cell Biol. 2021, 23, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.P.; Fletcher, A.G.; Baena-Lopez, L.A. Mechanisms and mechanics of cell competition in epithelia. Nat. Rev. Mol. Cell Biol. 2013, 14, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Nagata, R.; Igaki, T. Cell competition: Emerging mechanisms to eliminate neighbors. Dev. Growth Differ. 2018, 60, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Baillon, L.; Basler, K. Reflections on cell competition. Semin. Cell Dev. Biol. 2014, 32, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.E. Emerging mechanisms of cell competition. Nat. Rev. Genet. 2020, 21, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Fujita, Y. Cell competition in vertebrates—A key machinery for tissue homeostasis. Curr. Opin. Genet. Dev. 2022, 72, 15–21. [Google Scholar] [CrossRef]
- Villa Del Campo, C.; Torres, M. Changing the Rules of the Game: How Winners Become Losers during Oncogenic Cell Selection. Cell Stem Cell 2019, 25, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Martin, N.; Sierra, R.; Schimmang, T.; Villa Del Campo, C.; Torres, M. Myc is dispensable for cardiomyocyte development but rescues Mycn-deficient hearts through functional replacement and cell competition. Development 2019, 146, dev.170753. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Antoran, D.; Piedrafita, G.; Murai, K.; Ong, S.H.; Herms, A.; Frezza, C.; Jones, P.H. Outcompeting p53-Mutant Cells in the Normal Esophagus by Redox Manipulation. Cell Stem Cell 2019, 25, 329–341.e326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, M.M. Azot expression in the Drosophila gut modulates organismal lifespan. Commun. Integr. Biol. 2023, 16, 2156735. [Google Scholar] [CrossRef]
- Maheden, K.; Zhang, V.W.; Shakiba, N. The Field of Cell Competition Comes of Age: Semantics and Technological Synergy. Front. Cell Dev. Biol. 2022, 10, 891569. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.M.; Gonzalez-Gaitan, M. To fit or not to fit: Death decisions from morphogen fields. Trends Cell Biol. 2022, 33, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.M.; Seum, C.; Dubois, M.; Gonzalez-Gaitan, M. A role for Flower and cell death in controlling morphogen gradient scaling. Nat. Cell Biol. 2022, 24, 424–433. [Google Scholar] [CrossRef]
- Casas-Tinto, S.; Torres, M.; Moreno, E. The flower code and cancer development. Clin. Transl. Oncol. 2011, 13, 5–9. [Google Scholar] [CrossRef]
- Adachi-Yamada, T.; O’Connor, M.B. Morphogenetic apoptosis: A mechanism for correcting discontinuities in morphogen gradients. Dev. Biol. 2002, 251, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Basler, K.; Morata, G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 2002, 416, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Basler, K. dMyc transforms cells into super-competitors. Cell 2004, 117, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Adachi-Yamada, T.; O’Connor, M.B. Mechanisms for removal of developmentally abnormal cells: Cell competition and morphogenetic apoptosis. J. Biochem. 2004, 136, 13–17. [Google Scholar] [CrossRef]
- Baker, N.E.; Kale, A. Mutations in ribosomal proteins: Apoptosis, cell competition, and cancer. Mol. Cell Oncol. 2016, 3, e1029065. [Google Scholar] [CrossRef] [Green Version]
- de la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant, P. Myc, cell competition, and compensatory proliferation. Cancer Res. 2005, 65, 6485–6487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.P.; Kolahgar, G.; Gagliardi, M.; Piddini, E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell 2011, 21, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Akai, N.; Igaki, T.; Ohsawa, S. Wingless signaling regulates winner/loser status in Minute cell competition. Genes Cells 2018, 23, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Akieda, Y.; Ogamino, S.; Furuie, H.; Ishitani, S.; Akiyoshi, R.; Nogami, J.; Masuda, T.; Shimizu, N.; Ohkawa, Y.; Ishitani, T. Cell competition corrects noisy Wnt morphogen gradients to achieve robust patterning in the zebrafish embryo. Nat. Commun. 2019, 10, 4710. [Google Scholar] [CrossRef] [Green Version]
- Azubuike, U.F.; Tanner, K. Biophysical determinants of cancer organotropism. Trends Cancer 2022, 9, 188–197. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: Where are we now? Ther. Adv. Med. Oncol. 2018, 10, 1758834018757175. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, R.; Michalski, C.W.; Liu, B.; Liao, Q.; Kleeff, J. Surgery for synchronous and metachronous single-organ metastasis of pancreatic cancer: A SEER database analysis and systematic literature review. Sci. Rep. 2020, 10, 4444. [Google Scholar] [CrossRef] [Green Version]
- Gandaglia, G.; Karakiewicz, P.I.; Briganti, A.; Passoni, N.M.; Schiffmann, J.; Trudeau, V.; Graefen, M.; Montorsi, F.; Sun, M. Impact of the Site of Metastases on Survival in Patients with Metastatic Prostate Cancer. Eur. Urol. 2015, 68, 325–334. [Google Scholar] [CrossRef]
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Amersi, F.F.; Terando, A.M.; Goto, Y.; Scolyer, R.A.; Thompson, J.F.; Tran, A.N.; Faries, M.B.; Morton, D.L.; Hoon, D.S. Activation of CCR9/CCL25 in cutaneous melanoma mediates preferential metastasis to the small intestine. Clin. Cancer Res. 2008, 14, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Johnson-Holiday, C.; Singh, R.; Johnson, E.; Singh, S.; Stockard, C.R.; Grizzle, W.E.; Lillard, J.W., Jr. CCL25 mediates migration, invasion and matrix metalloproteinase expression by breast cancer cells in a CCR9-dependent fashion. Int. J. Oncol. 2011, 38, 1279–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letsch, A.; Keilholz, U.; Schadendorf, D.; Assfalg, G.; Asemissen, A.M.; Thiel, E.; Scheibenbogen, C. Functional CCR9 expression is associated with small intestinal metastasis. J. Investig. Dermatol. 2004, 122, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirandola, L.; Chiriva-Internati, M.; Montagna, D.; Locatelli, F.; Zecca, M.; Ranzani, M.; Basile, A.; Locati, M.; Cobos, E.; Kast, W.M.; et al. Notch1 regulates chemotaxis and proliferation by controlling the CC-chemokine receptors 5 and 9 in T cell acute lymphoblastic leukaemia. J. Pathol. 2012, 226, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Qiuping, Z.; Qun, L.; Chunsong, H.; Xiaolian, Z.; Baojun, H.; Mingzhen, Y.; Chengming, L.; Jinshen, H.; Qingping, G.; Kejian, Z.; et al. Selectively increased expression and functions of chemokine receptor CCR9 on CD4+ T cells from patients with T-cell lineage acute lymphocytic leukemia. Cancer Res. 2003, 63, 6469–6477. [Google Scholar]
- Singh, S.; Singh, U.P.; Stiles, J.K.; Grizzle, W.E.; Lillard, J.W., Jr. Expression and functional role of CCR9 in prostate cancer cell migration and invasion. Clin. Cancer Res. 2004, 10, 8743–8750. [Google Scholar] [CrossRef] [Green Version]
- van den Oord, J. The CCR9-CCL25 axis mediates melanoma metastasis to the small intestine. Nat. Clin. Pract. Oncol. 2008, 5, 440–441. [Google Scholar] [CrossRef]
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A. Paget’s “Seed and Soil” Theory of Cancer Metastasis: An Idea Whose Time has Come. Adv. Anat. Pathol. 2019, 26, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.A.W. On Growth and Form; Cambridge University Press: Cambridge, UK, 1917. [Google Scholar]
- Hamaratoglu, F.; de Lachapelle, A.M.; Pyrowolakis, G.; Bergmann, S.; Affolter, M. Dpp signaling activity requires Pentagone to scale with tissue size in the growing Drosophila wing imaginal disc. PLoS Biol. 2011, 9, e1001182. [Google Scholar] [CrossRef] [Green Version]
- Stapornwongkul, K.S.; de Gennes, M.; Cocconi, L.; Salbreux, G.; Vincent, J.P. Patterning and growth control in vivo by an engineered GFP gradient. Science 2020, 370, 321–327. [Google Scholar] [CrossRef]
- Martin, F.A.; Perez-Garijo, A.; Moreno, E.; Morata, G. The brinker gradient controls wing growth in Drosophila. Development 2004, 131, 4921–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellen, D.; Burke, R.; Struhl, G.; Basler, K. Direct and long-range action of a DPP morphogen gradient. Cell 1996, 85, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartlick, O.; Mumcu, P.; Kicheva, A.; Bittig, T.; Seum, C.; Julicher, F.; Gonzalez-Gaitan, M. Dynamics of Dpp Signaling and Proliferation Control. Science 2011, 331, 1154–1159. [Google Scholar] [CrossRef]
- Zecca, M.; Struhl, G. A unified mechanism for the control of Drosophila wing growth by the morphogens Decapentaplegic and Wingless. PLoS Biol. 2021, 19, e3001111. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Schaefer, J.V.; Mii, Y.; Hori, Y.; Bieli, D.; Taira, M.; Pluckthun, A.; Affolter, M. Asymmetric requirement of Dpp/BMP morphogen dispersal in the Drosophila wing disc. Nat. Commun. 2021, 12, 6435. [Google Scholar] [CrossRef]
- Harmansa, S.; Hamaratoglu, F.; Affolter, M.; Caussinus, E. Dpp spreading is required for medial but not for lateral wing disc growth. Nature 2015, 527, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Hamaratoglu, F.; Affolter, M.; Pyrowolakis, G. Dpp/BMP signaling in flies: From molecules to biology. Semin. Cell Dev. Biol. 2014, 32, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.; Kamimura, K.; Firkus, C.; Takeo, S.; Shimmi, O.; Nakato, H. Dally regulates Dpp morphogen gradient formation by stabilizing Dpp on the cell surface. Dev. Biol. 2008, 313, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuilleumier, R.; Springhorn, A.; Patterson, L.; Koidl, S.; Hammerschmidt, M.; Affolter, M.; Pyrowolakis, G. Control of Dpp morphogen signalling by a secreted feedback regulator. Nat. Cell Biol. 2010, 12, 611–617. [Google Scholar] [CrossRef]
- Simon, N.; Safyan, A.; Pyrowolakis, G.; Matsuda, S. Dally is not essential for Dpp spreading or internalization but for Dpp stability by antagonizing Tkv-mediated Dpp internalization. bioRxiv 2023. [Google Scholar] [CrossRef]
- Norman, M.; Vuilleumier, R.; Springhorn, A.; Gawlik, J.; Pyrowolakis, G. Pentagone internalises glypicans to fine-tune multiple signalling pathways. Elife 2016, 5, e13301. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, D.; Pyrowolakis, G.; Barkai, N.; Shilo, B.Z. Expansion-repression mechanism for scaling the Dpp activation gradient in Drosophila wing imaginal discs. Curr. Biol. 2011, 21, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bely, A.E.; Nyberg, K.G. Evolution of animal regeneration: Re-emergence of a field. Trends Ecol. Evol. 2010, 25, 161–170. [Google Scholar] [CrossRef]
- Maden, M. The evolution of regeneration—Where does that leave mammals? Int. J. Dev. Biol. 2018, 62, 369–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.G.; Aboobaker, A.A. EvoRegen in animals: Time to uncover deep conservation or convergence of adult stem cell evolution and regenerative processes. Dev. Biol. 2018, 433, 118–131. [Google Scholar] [CrossRef]
- Mubbunu, L.; Bowa, K.; Petrenko, V.; Silitongo, M. Correlation of Internal Organ Weights with Body Weight and Body Height in Normal Adult Zambians: A Case Study of Ndola Teaching Hospital. Anat. Res. Int. 2018, 2018, 4687538. [Google Scholar] [CrossRef]
- Vaibhav, V.; Meshram, R.; Shukla, P.K.; Kalonia, T.; Bhute, A.R. A Preliminary Study of Organ Weight After Histological Exclusion of Abnormality During Autopsy in the Adult Population of Uttarakhand, India. Cureus 2022, 14, e27044. [Google Scholar] [CrossRef]
- Greif, J.M.; Pezzi, C.M.; Klimberg, V.S.; Bailey, L.; Zuraek, M. Gender differences in breast cancer: Analysis of 13,000 breast cancers in men from the National Cancer Data Base. Ann. Surg. Oncol. 2012, 19, 3199–3204. [Google Scholar] [CrossRef]
- Li, H.; Wei, Z.; Wang, C.; Chen, W.; He, Y.; Zhang, C. Gender Differences in Gastric Cancer Survival: 99,922 Cases Based on the SEER Database. J. Gastrointest. Surg. 2020, 24, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Blackford, A.L.; Dal Molin, M.; Wolfgang, C.L.; Goggins, M. Time to progression of pancreatic ductal adenocarcinoma from low-to-high tumour stages. Gut 2015, 64, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Li, W.; Cai, S.; Yang, C.; Liu, Y.; Lin, Z. Large tumor size is a poor prognostic factor of gastric cancer with signet ring cell: Results from the surveillance, epidemiology, and end results database. Medicine 2019, 98, e17367. [Google Scholar] [CrossRef]
- Kasangian, A.A.; Gherardi, G.; Biagioli, E.; Torri, V.; Moretti, A.; Bernardin, E.; Cordovana, A.; Farina, G.; Bramati, A.; Piva, S.; et al. The prognostic role of tumor size in early breast cancer in the era of molecular biology. PLoS ONE 2017, 12, e0189127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.C., Jr.; Garg, A.; Goswitz, J.; Cheng, E.Y.; Clohisy, D.R.; Dusenbery, K. Synovial sarcoma. Large size predicts poor outcome. Clin. Orthop. Relat. Res. 2000, 373, 18–24. [Google Scholar] [CrossRef]
- Wunderlich, S.; Haase, A.; Merkert, S.; Jahn, K.; Deest, M.; Frieling, H.; Glage, S.; Korte, W.; Martens, A.; Kirschning, A.; et al. Targeted biallelic integration of an inducible Caspase 9 suicide gene in iPSCs for safer therapies. Mol. Ther. Methods Clin. Dev. 2022, 26, 84–94. [Google Scholar] [CrossRef]
- Tian, Y.; Zhao, S.; Zheng, J.; Li, Z.; Hou, C.; Qi, X.; Kong, D.; Zhang, J.; Huang, X. A stereological study of 3D printed tissues engineered from rat vaginas. Ann. Transl. Med. 2020, 8, 1490. [Google Scholar] [CrossRef]
- Holt, P.G.; Degebrodt, A.; Venaille, T.; O’Leary, C.; Krska, K.; Flexman, J.; Farrell, H.; Shellam, G.; Young, P.; Penhale, J.; et al. Preparation of interstitial lung cells by enzymatic digestion of tissue slices: Preliminary characterization by morphology and performance in functional assays. Immunology 1985, 54, 139–147. [Google Scholar] [PubMed]
- Del Monte, U. Does the cell number 10(9) still really fit one gram of tumor tissue? Cell Cycle 2009, 8, 505–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affolter, M.; Basler, K. The Decapentaplegic morphogen gradient: From pattern formation to growth regulation. Nat. Rev. Genet. 2007, 8, 663–674. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merino, M.M.; Garcia-Sanz, J.A. Stemming Tumoral Growth: A Matter of Grotesque Organogenesis. Cells 2023, 12, 872. https://doi.org/10.3390/cells12060872
Merino MM, Garcia-Sanz JA. Stemming Tumoral Growth: A Matter of Grotesque Organogenesis. Cells. 2023; 12(6):872. https://doi.org/10.3390/cells12060872
Chicago/Turabian StyleMerino, Marisa M., and Jose A. Garcia-Sanz. 2023. "Stemming Tumoral Growth: A Matter of Grotesque Organogenesis" Cells 12, no. 6: 872. https://doi.org/10.3390/cells12060872