
Pathogenic Microglia Orchestrate Neurotoxic Properties of Eomes-Expressing Helper T Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. EAE Induction and Scoring
2.3. Treatment of Animals
2.4. Cell Isolation
2.5. BMDC Culture
2.6. Calcium Flux Assay
2.7. Flow Cytometry
2.8. Cell Sorting
2.9. Mouse Vβ TCR Analysis
2.10. Real Time qPCR
2.11. Microarray Analysis
2.12. Single-Cell Capture, Imaging, and qPCR Analysis
2.13. Immunohistochemistry
2.14. In Vitro Translation Protein Expression
2.15. ORF-1 Peptide Library
2.16. Statistical Analysis
3. Results
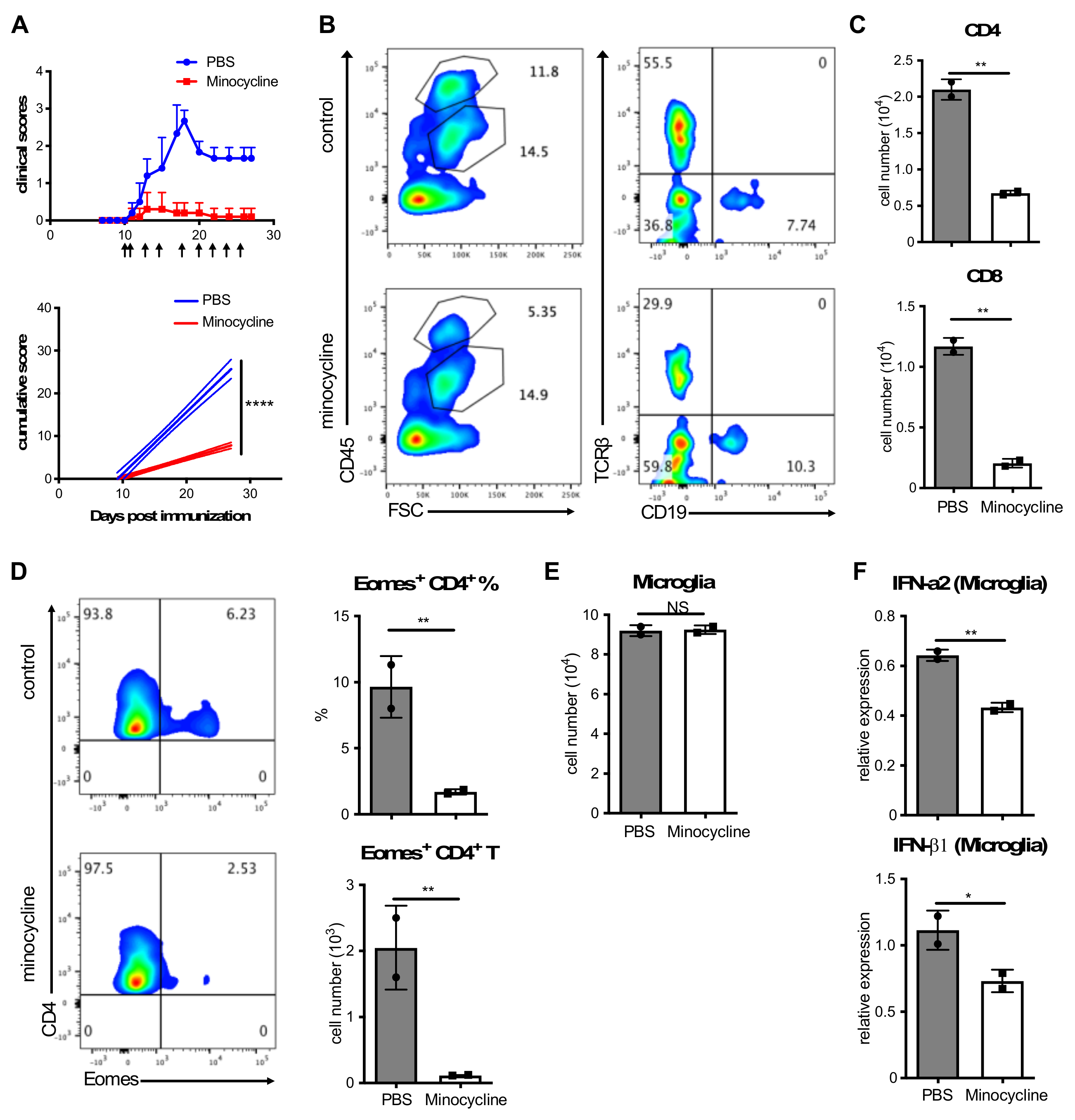
3.1. Inhibition of Microglia Activation Attenuates Clinical EAE
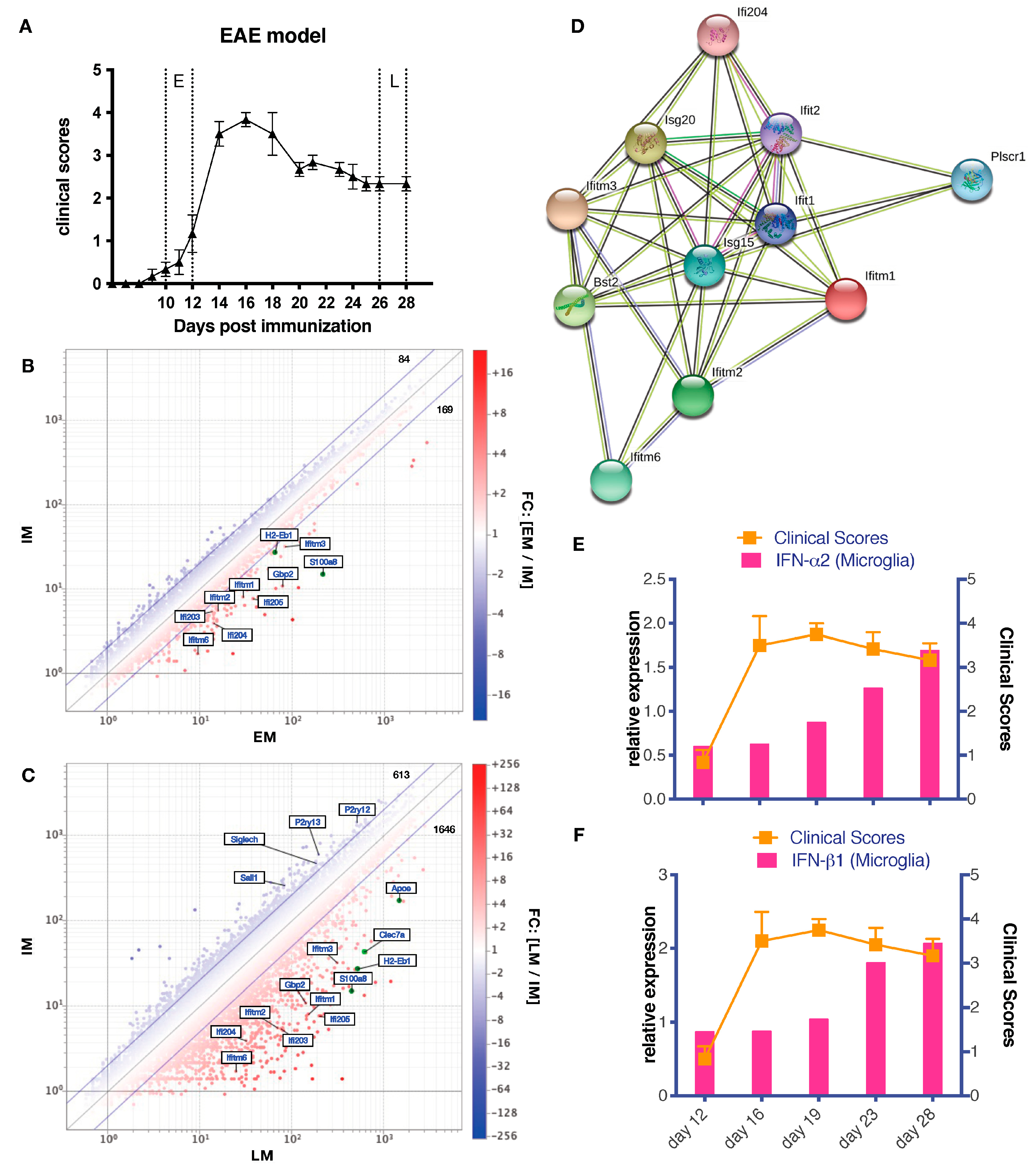
3.2. Differential Gene Expression in Late EAE Microglia
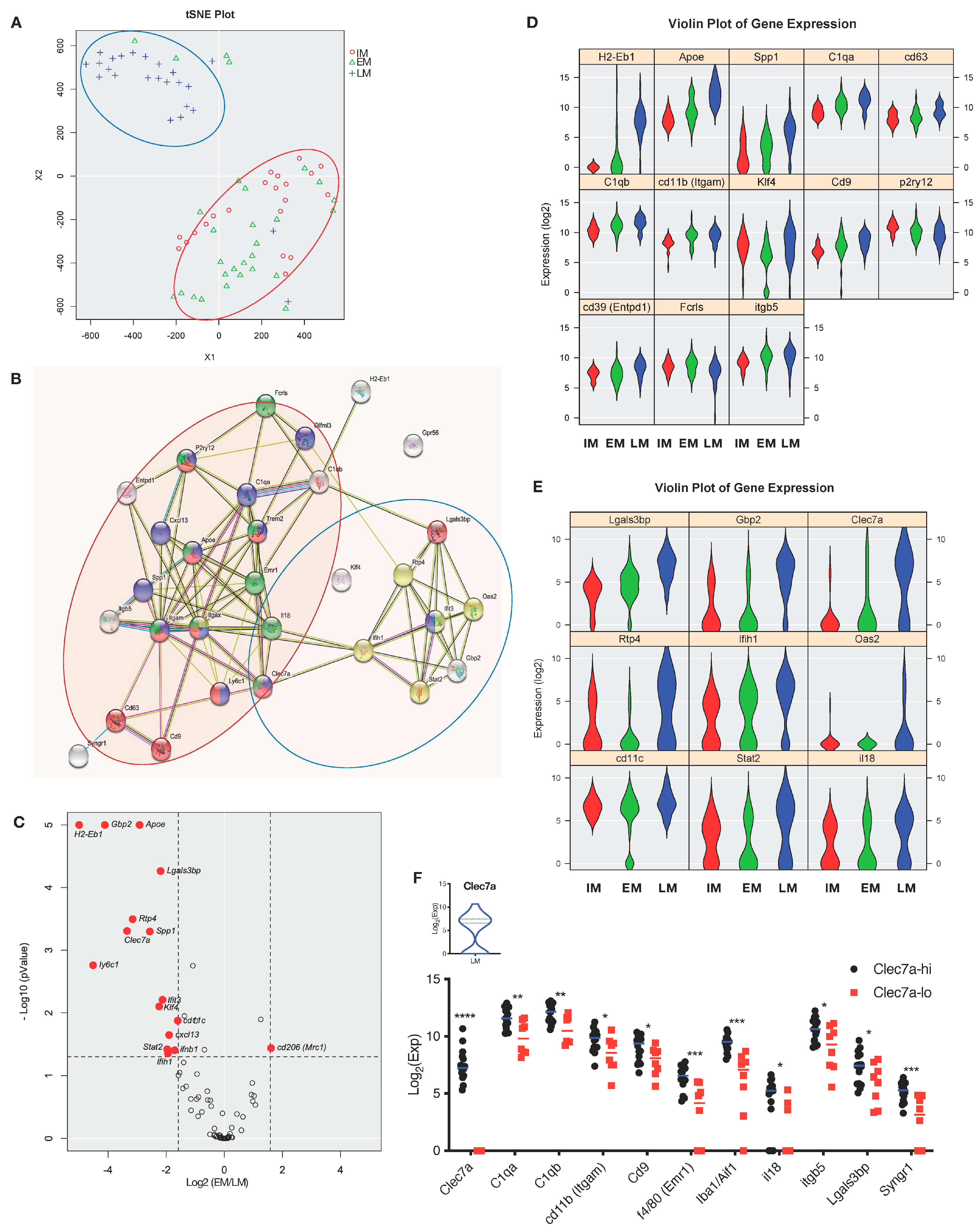
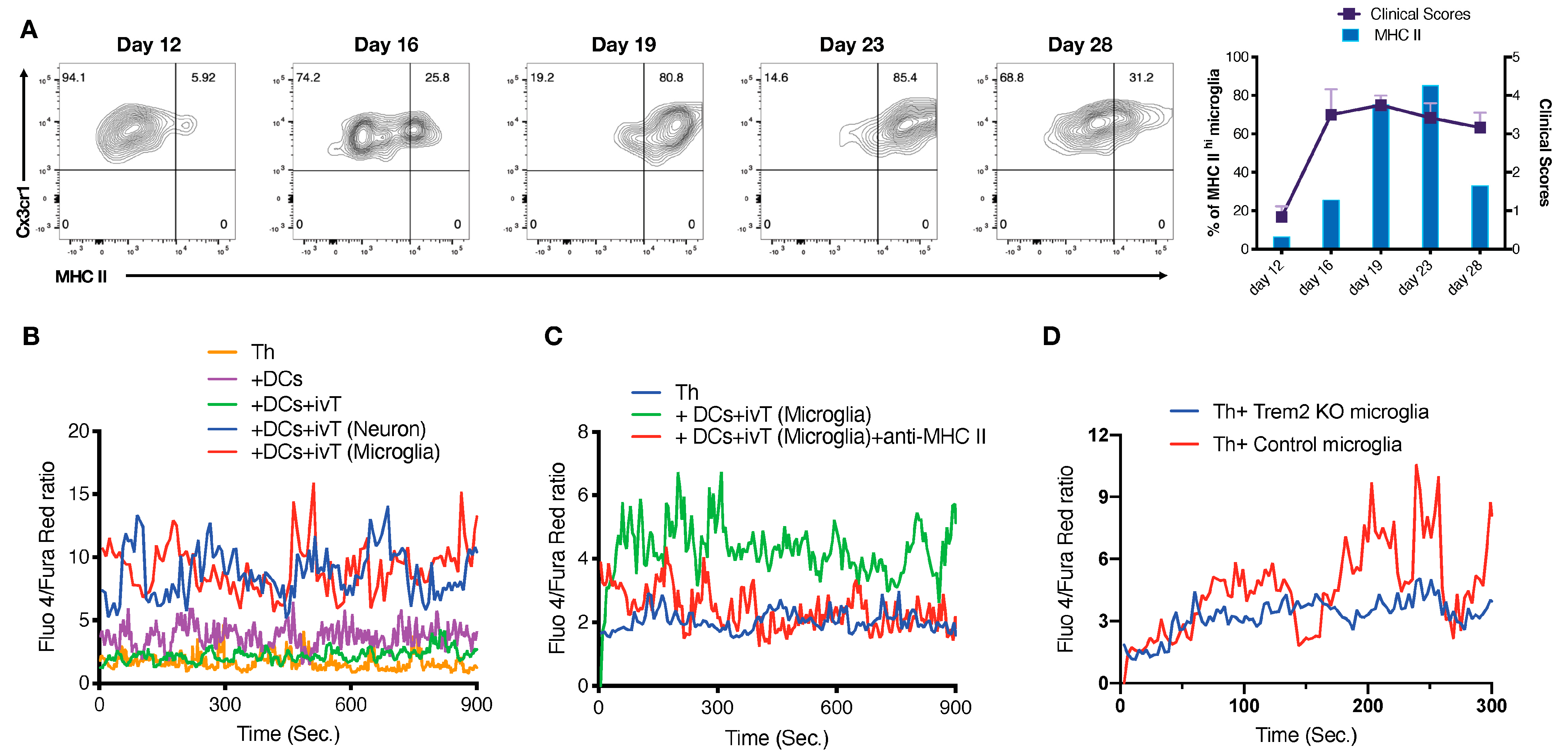
3.3. Functional Heterogeneity of EAE-Associated Microglia
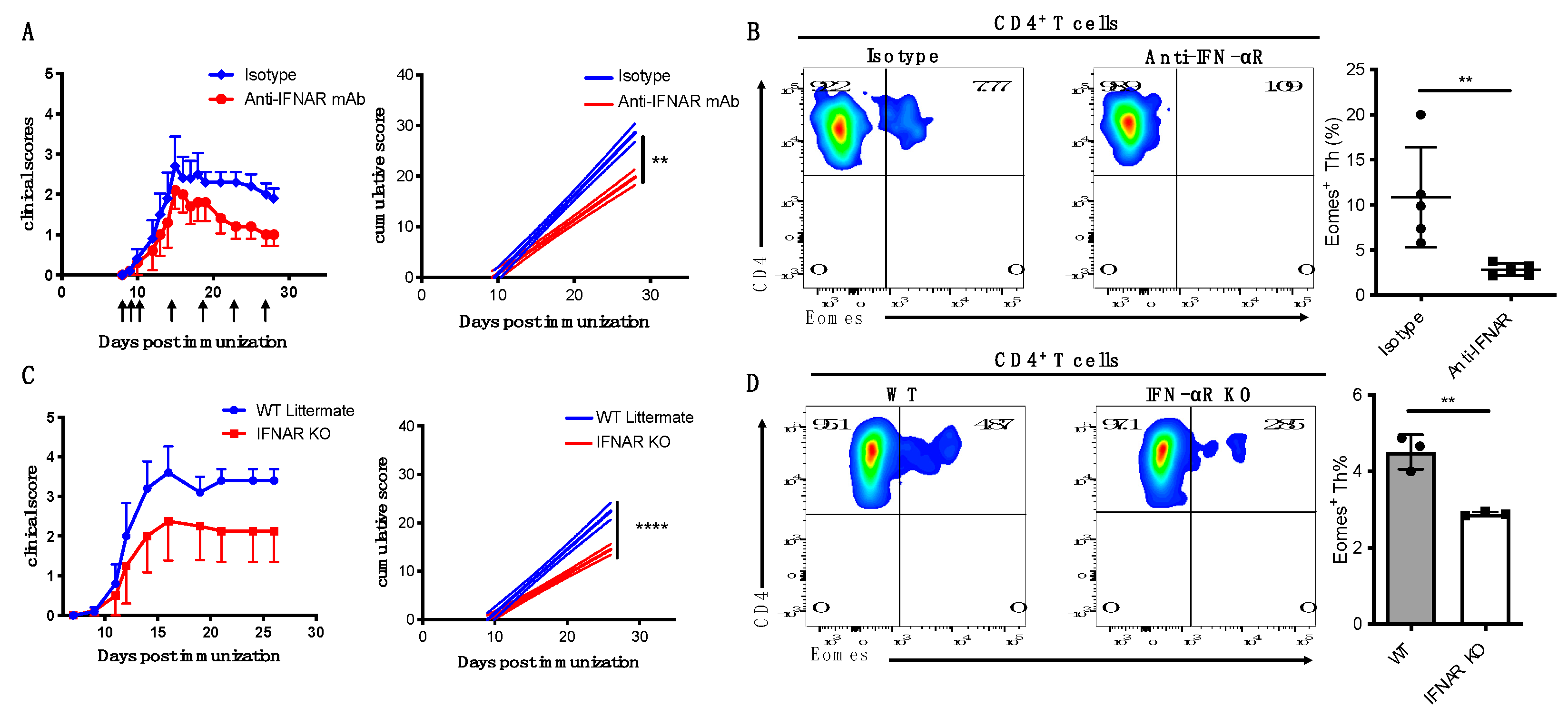
3.4. IFN-I Is Critical for Induction of Eomes+ Th Cells and Development of Late EAE
3.5. CNS Antigens Act as Trigger for Pathogenesis in Late EAE Disease
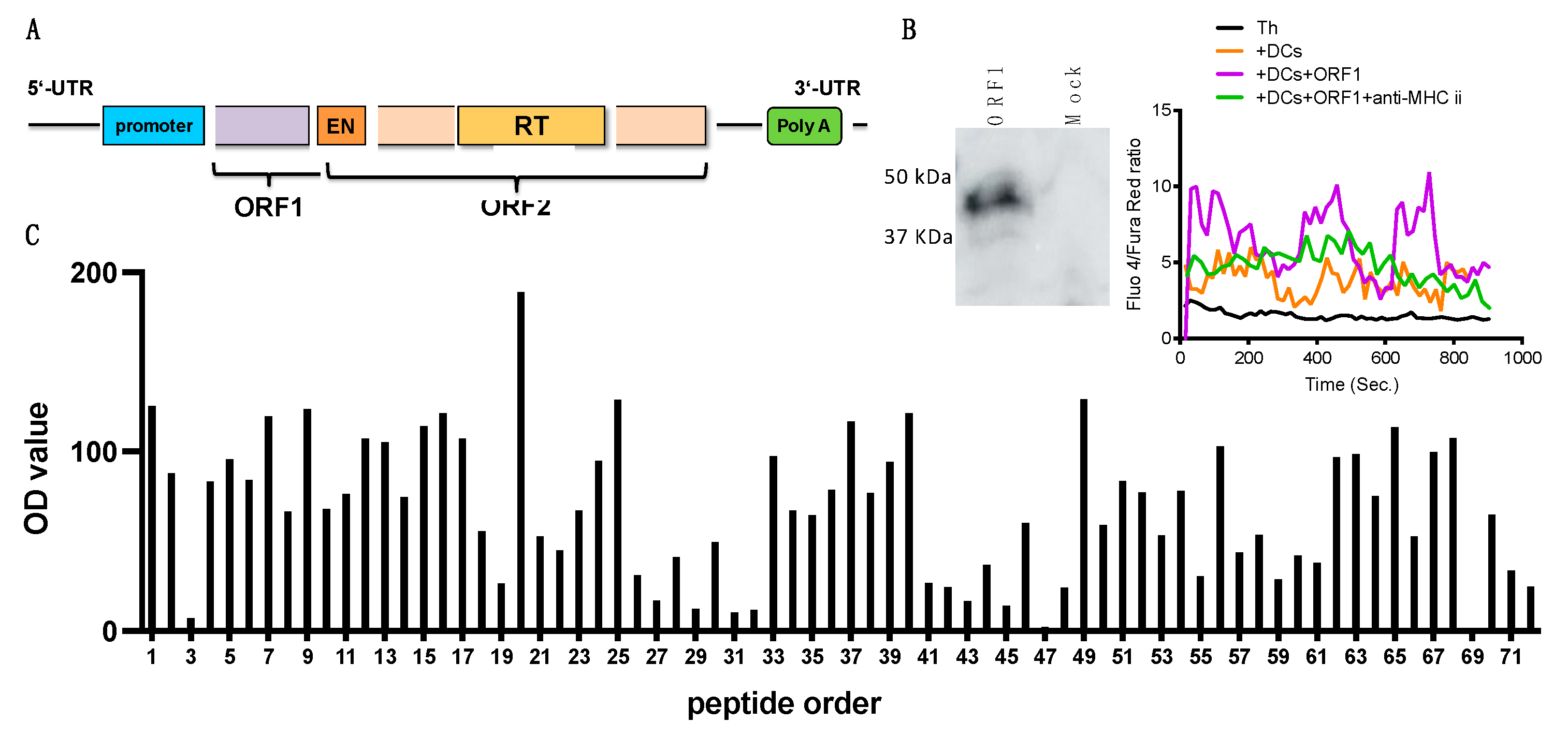
3.6. ORF-1, an Encoded Protein of L1 Retrotransposon May Contribute to CNS Th Cell Activation
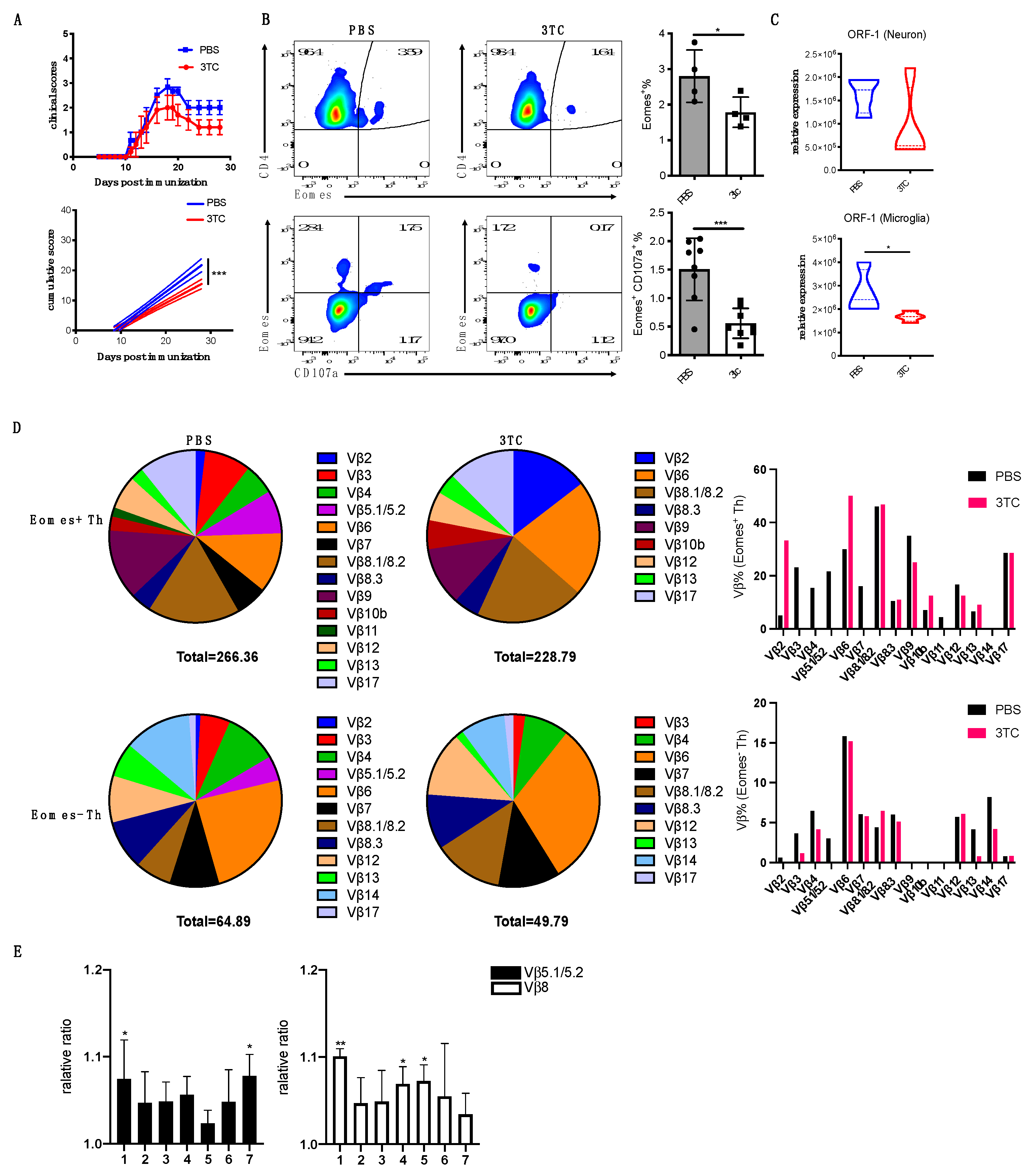
3.7. Blockade of L1 Activation Ameliorates Late EAE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Sun, W.Y.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Guardia-Laguarta, C.; Schon, E.A.; Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019, 129, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, R.; Samokhina, E.; Rossetti, I.; Morley, J.W.; Buskila, Y. Neuromodulation of Glial Function during Neurodegeneration. Front. Cell Neurosci. 2020, 14, 278. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Hakim-Mishnaevski, K.; Flint-Brodsly, N.; Shklyar, B.; Levy-Adam, F.; Kurant, E. Glial Phagocytic Receptors Promote Neuronal Loss in Adult Drosophila Brain. Cell Rep. 2019, 29, 1438–1448.e3. [Google Scholar] [CrossRef]
- Werneburg, S.; Jung, J.; Kunjamma, R.B.; Ha, S.K.; Luciano, N.J.; Willis, C.M.; Gao, G.; Biscola, N.P.; Havton, L.A.; Crocker, S.J.; et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2020, 52, 167–182.e7. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Cho, C.E.; Damle, S.S.; Wancewicz, E.V.; Mukhopadhyay, S.; Hart, C.E.; Mazur, C.; Swayze, E.E.; Kamme, F. A modular analysis of microglia gene expression, insights into the aged phenotype. BMC Genom. 2019, 20, 164. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 2021, 218, e20202717. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Correale, J.; Marrodan, M.; Ysrraelit, M.C. Mechanisms of Neurodegeneration and Axonal Dysfunction in Progressive Multiple Sclerosis. Biomedicines 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Raveney, B.J.; Oki, S.; Hohjoh, H.; Nakamura, M.; Sato, W.; Murata, M.; Yamamura, T. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nat. Commun. 2015, 6, 8437. [Google Scholar] [CrossRef] [Green Version]
- Raveney, B.J.E.; Sato, W.; Takewaki, D.; Zhang, C.; Kanazawa, T.; Lin, Y.; Okamoto, T.; Araki, M.; Kimura, Y.; Sato, N.; et al. Involvement of cytotoxic Eomes-expressing CD4+ T cells in secondary progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2021818118. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, F.; Zhang, C.; Hohjoh, H.; Raveney, B.; Yamamura, T.; Hayashi, N.; Oki, S. Immune-mediated neurodegenerative trait provoked by multimodal derepression of long-interspersed nuclear element-1. iScience 2022, 25, 104278. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Reder, A.T. Therapeutic role of beta-interferons in multiple sclerosis. Pharmacol. Ther. 2006, 110, 35–56. [Google Scholar] [CrossRef]
- Crow, Y.J.; Manel, N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef]
- Goldmann, T.; Zeller, N.; Raasch, J.; Kierdorf, K.; Frenzel, K.; Ketscher, L.; Basters, A.; Staszewski, O.; Brendecke, S.M.; Spiess, A.; et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015, 34, 1612–1629. [Google Scholar] [CrossRef] [Green Version]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2017, 8, 1905. [Google Scholar] [CrossRef] [Green Version]
- Wlodarczyk, A.; Lobner, M.; Cedile, O.; Owens, T. Comparison of microglia and infiltrating CD11c(+) cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflamm. 2014, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Bundo, M.; Toyoshima, M.; Okada, Y.; Akamatsu, W.; Ueda, J.; Nemoto-Miyauchi, T.; Sunaga, F.; Toritsuka, M.; Ikawa, D.; Kakita, A.; et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 2014, 81, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohjoh, H.; Singer, M.F. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 1996, 15, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Hohjoh, H.; Singer, M.F. Sequence-specific single-strand RNA binding protein encoded by the human LINE-1 retrotransposon. EMBO J. 1997, 16, 6034–6043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Raveney, B.J.E.; Hohjoh, H.; Tomi, C.; Oki, S.; Yamamura, T. Extrapituitary prolactin promotes generation of Eomes-positive helper T cells mediating neuroinflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 21131–21139. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Xu, F.; Previti, M.L.; Davis, J.; Grande, A.M.; Robinson, J.K.; Van Nostrand, W.E. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J. Neurosci. 2007, 27, 3057–3063. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef] [Green Version]
- Del Fresno, C.; Soulat, D.; Roth, S.; Blazek, K.; Udalova, I.; Sancho, D.; Ruland, J.; Ardavin, C. Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 2013, 38, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Schmidt, H.; Mildner, A.; Knobeloch, K.P.; Hanisch, U.K.; Raasch, J.; Merkler, D.; Detje, C.; Gutcher, I.; Mages, J.; et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 2008, 28, 675–686. [Google Scholar] [CrossRef]
- Kocur, M.; Schneider, R.; Pulm, A.K.; Bauer, J.; Kropp, S.; Gliem, M.; Ingwersen, J.; Goebels, N.; Alferink, J.; Prozorovski, T.; et al. IFNbeta secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol. Commun. 2015, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.R.; Morell, S.; Faulkner, G.J. L1 retrotransposons and somatic mosaicism in the brain. Annu. Rev. Genet. 2014, 48, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Banuelos-Sanchez, G.; Sanchez, L.; Benitez-Guijarro, M.; Sanchez-Carnerero, V.; Salvador-Palomeque, C.; Tristan-Ramos, P.; Benkaddour-Boumzaouad, M.; Morell, S.; Garcia-Puche, J.L.; Heras, S.R.; et al. Synthesis and Characterization of Specific Reverse Transcriptase Inhibitors for Mammalian LINE-1 Retrotransposons. Cell Chem. Biol. 2019, 26, 1095–1109.e14. [Google Scholar] [CrossRef]
- Shimada, A.; Hasegawa-Ishii, S. Histological Architecture Underlying Brain-Immune Cell-Cell Interactions and the Cerebral Response to Systemic Inflammation. Front. Immunol. 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, N.; Fujita, Y.; Temmoku, J.; Furuya, M.Y.; Asano, T.; Sato, S.; Matsumoto, H.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; et al. Galectin-9 as a biomarker for disease activity in systemic lupus erythematosus. PLoS ONE 2020, 15, e0227069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Ochi, A.; Heindel, D.W.; Lee, K.B.; Zambirinis, C.P.; Pandian, G.S.B.; Savadkar, S.; et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat. Med. 2017, 23, 556–567. [Google Scholar] [CrossRef]
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef]
- Schmeisser, H.; Fey, S.B.; Horowitz, J.; Fischer, E.R.; Balinsky, C.A.; Miyake, K.; Bekisz, J.; Snow, A.L.; Zoon, K.C. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 2013, 9, 683–696. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Minati, R.; Perreault, C.; Thibault, P. A Roadmap Toward the Definition of Actionable Tumor-Specific Antigens. Front. Immunol. 2020, 11, 583287. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef] [Green Version]
- Mavragani, C.P.; Sagalovskiy, I.; Guo, Q.; Nezos, A.; Kapsogeorgou, E.K.; Lu, P.; Zhou, J.L.; Kirou, K.A.; Seshan, S.V.; Moutsopoulos, H.M.; et al. Expression of Long Interspersed Nuclear Element 1 Retroelements and Induction of Type I Interferon in Patients With Systemic Autoimmune Disease. Arthritis Rheumatol. 2016, 68, 2686–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez-Guijarro, M.; Lopez-Ruiz, C.; Tarnauskaite, Z.; Murina, O.; Mian Mohammad, M.; Williams, T.C.; Fluteau, A.; Sanchez, L.; Vilar-Astasio, R.; Garcia-Canadas, M.; et al. RNase H2, mutated in Aicardi-Goutieres syndrome, promotes LINE-1 retrotransposition. EMBO J. 2018, 37, e98506. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.C.; Sanchez, L.; Schaughency, P.M.; Rubio-Roldan, A.; Choi, J.A.; Planet, E.; Batra, R.; Turelli, P.; Trono, D.; Ostrow, L.W.; et al. Properties of LINE-1 proteins and repeat element expression in the context of amyotrophic lateral sclerosis. Mob. DNA 2018, 9, 35. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Carbone, C.J.; Katlinskaya, Y.V.; Zheng, H.; Zheng, K.; Luo, M.; Wang, P.J.; Greenberg, R.A.; Fuchs, S.Y. Type I interferon controls propagation of long interspersed element-1. J. Biol. Chem. 2015, 290, 10191–10199. [Google Scholar] [CrossRef] [Green Version]
- Terry, D.M.; Devine, S.E. Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders. Front. Genet. 2019, 10, 1244. [Google Scholar] [CrossRef] [Green Version]
- Carter, V.; LaCava, J.; Taylor, M.S.; Liang, S.Y.; Mustelin, C.; Ukadike, K.C.; Bengtsson, A.; Lood, C.; Mustelin, T. High Prevalence and Disease Correlation of Autoantibodies Against p40 Encoded by Long Interspersed Nuclear Elements in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2020, 72, 89–99. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Raveney, B.; Takahashi, F.; Yeh, T.-w.; Hohjoh, H.; Yamamura, T.; Oki, S. Pathogenic Microglia Orchestrate Neurotoxic Properties of Eomes-Expressing Helper T Cells. Cells 2023, 12, 868. https://doi.org/10.3390/cells12060868
Zhang C, Raveney B, Takahashi F, Yeh T-w, Hohjoh H, Yamamura T, Oki S. Pathogenic Microglia Orchestrate Neurotoxic Properties of Eomes-Expressing Helper T Cells. Cells. 2023; 12(6):868. https://doi.org/10.3390/cells12060868
Chicago/Turabian StyleZhang, Chenyang, Ben Raveney, Fumio Takahashi, Tzu-wen Yeh, Hirohiko Hohjoh, Takashi Yamamura, and Shinji Oki. 2023. "Pathogenic Microglia Orchestrate Neurotoxic Properties of Eomes-Expressing Helper T Cells" Cells 12, no. 6: 868. https://doi.org/10.3390/cells12060868