
Revealing the Heterogeneity of the Tumor Ecosystem of Cholangiocarcinoma through Single-Cell Transcriptomics


Abstract
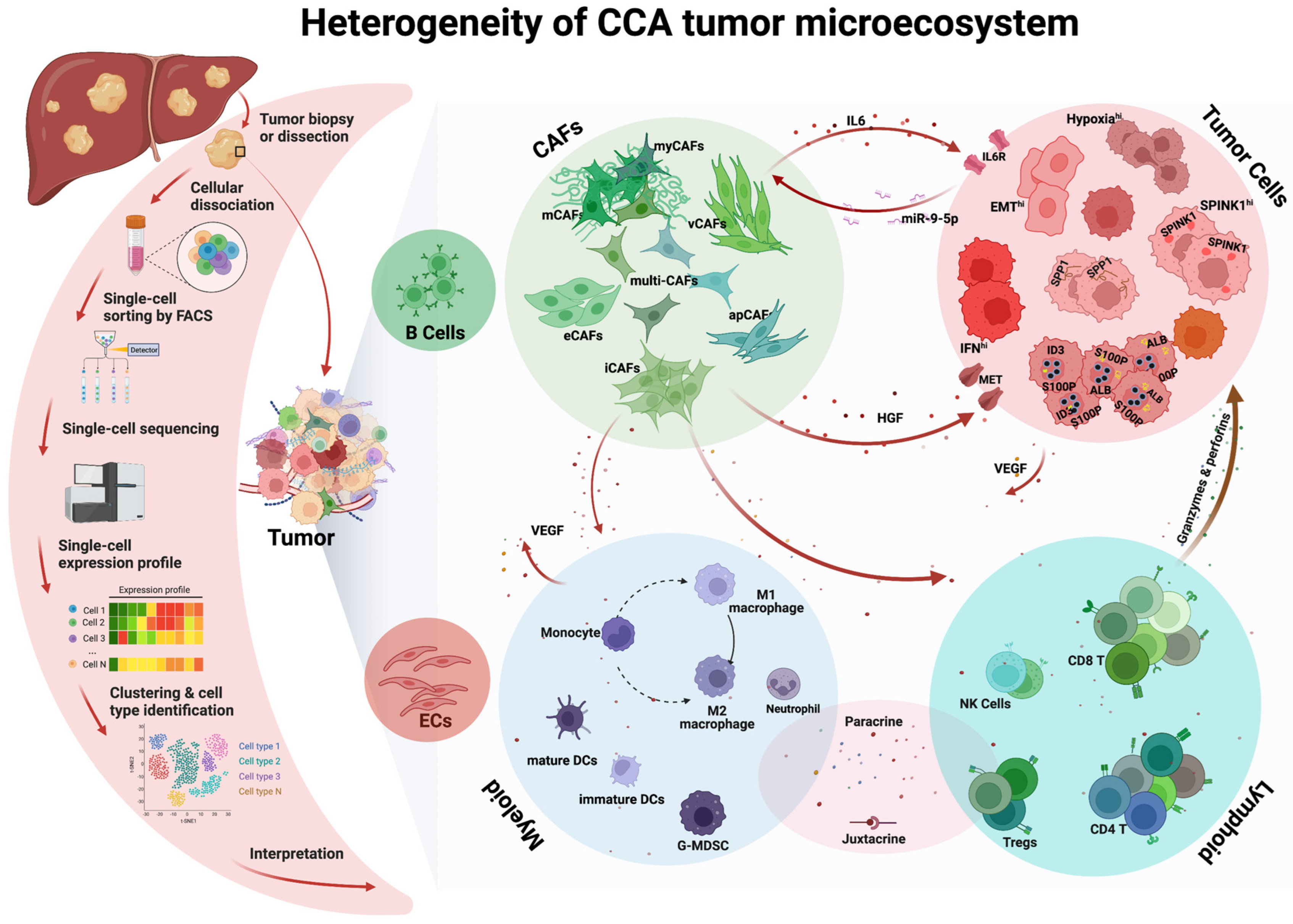
:1. Introduction
2. Heterogeneity and Plasticity of CCA Tumor Cells
3. Heterogeneity of CCA Cancer-Associated Fibroblasts (CAFs)
4. Heterogeneity of CCA Immune Cells
4.1. Lymphoid Compartment
4.2. Myeloid Compartment
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef]
- Van Dyke, A.L.; Shiels, M.S.; Jones, G.S.; Pfeiffer, R.M.; Petrick, J.L.; Beebe-Dimmer, J.L.; Koshiol, J. Biliary tract cancer incidence and trends in the United States by demographic group, 1999–2013. Cancer 2019, 125, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Fletcher, P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Long-Term Outcomes and Exploratory Analyses of the Randomized Phase III BILCAP Study. J. Clin. Oncol. 2022, 40, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Coelen, R.J.; Gaspersz, M.P.; Labeur, T.A.; van Vugt, J.L.; van Dieren, S.; Willemssen, F.E.; Nio, C.Y.; Ijzermans, J.N.; Klümpen, H.-J.; Koerkamp, B.G.; et al. Validation of the Mayo Clinic Staging System in Determining Prognoses of Patients with Perihilar Cholangiocarcinoma. Clin. Gastroenterol. Hepatol. 2017, 15, 1930–1939.e3. [Google Scholar] [CrossRef]
- Murad, S.D.; Kim, W.R.; Harnois, D.M.; Douglas, D.D.; Burton, J.; Kulik, L.M.; Botha, J.F.; Mezrich, J.D.; Chapman, W.C.; Schwartz, J.J.; et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012, 143, 88–98.e3. [Google Scholar] [CrossRef] [Green Version]
- Lunsford, K.E.; Javle, M.; Heyne, K.; Shroff, R.T.; Abdel-Wahab, R.; Gupta, N.; Mobley, C.M.; Saharia, A.; Victor, D.W.; Nguyen, D.T.; et al. Liver transplantation for locally advanced intrahepatic cholangiocarcinoma treated with neoadjuvant therapy: A prospective case-series. Lancet Gastroenterol. Hepatol. 2018, 3, 337–348. [Google Scholar] [CrossRef]
- Twohig, P.; Peeraphatdit, T.B.; Mukherjee, S. Current status of liver transplantation for cholangiocarcinoma. World J. Gastrointest. Surg. 2022, 14, 6265. [Google Scholar] [CrossRef]
- Sapisochin, G.; Ivanics, T.; Heimbach, J. Liver Transplantation for Intrahepatic Cholangiocarcinoma: Ready for Prime Time? Hepatology 2022, 75, 455–472. [Google Scholar] [CrossRef]
- Manzia, T.M.; Parente, A.; Lenci, I.; Sensi, B.; Milana, M.; Gazia, C.; Signorello, A.; Angelico, R.; Grassi, G.; Tisone, G.; et al. Moving forward in the treatment of cholangiocarcinoma. World J. Gastrointest. Oncol. 2021, 13, 1939–1955. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Gorgen, A.; Roayaie, S.; Droz Dit Busset, M.; Sapisochin, G. Liver resection and transplantation for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 364–377. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; McGrath, N.A.; Monge Bonilla, C.; Fu, J. Systemic treatment options for advanced biliary tract carcinoma. J. Gastroenterol. 2020, 55, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Guthrie, K.A.; Scott, A.J.; Borad, M.J.; Goff, L.W.; Matin, K.; Mahipal, A.; Kalyan, A.; Javle, M.M.; Aghajanian, C.; et al. SWOG 1815: A phase III randomized trial of gemcitabine, cisplatin, and nab-paclitaxel versus gemcitabine and cisplatin in newly diagnosed, advanced biliary tract cancers. J. Clin. Oncol. 2023, 41 (Suppl. S4), LBA490. [Google Scholar]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Lee, M.A.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): A phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Kim, K.-P.; Jeong, J.H.; Kim, I.; Kang, M.J.; Cheon, J.; Kang, B.W.; Ryu, H.; Lee, J.S.; Kim, K.W.; et al. Liposomal irinotecan plus fluorouracil and leucovorin versus fluorouracil and leucovorin for metastatic biliary tract cancer after progression on gemcitabine plus cisplatin (NIFTY): A multicentre, open-label, randomised, phase 2b study. Lancet Oncol. 2021, 22, 1560–1572. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Javle, M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Weiss, K.H.; Waldschmidt, D.-T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.; et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: Mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol. 2021, 6, 803–815. [Google Scholar] [CrossRef]
- Goyal, L.; Bahleda, R.; Furuse, J.; Valle, J.W.; Moehler, M.H.; Oh, D.-Y.; Chang, H.-M.; Kelley, R.K.; Javle, M.M.; Borad, M.J.; et al. FOENIX-101: A phase II trial of TAS-120 in patients with intrahepatic cholangiocarcinoma harboring FGFR2 gene rearrangements. J. Clin. Oncol. 2019, 37 (Suppl. S4), TPS468. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients with Advanced Cholangiocarcinoma with IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef]
- Greten, T.F.; Schwabe, R.; Bardeesy, N.; Ma, L.; Goyal, L.; Kelley, R.K.; Wang, X.W. Immunology and immunotherapy of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, M.A.; Kepecs, B.; Torres-Martin, M.; Bramel, E.R.; Haber, P.K.; Merritt, E.; Rialdi, A.; Param, N.J.; Maeda, M.; Lindblad, K.E.; et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 2022, 72, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Finotello, F.; Trajanoski, Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol. Immunother. 2018, 67, 1031–1040. [Google Scholar] [CrossRef]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef]
- Ma, L.; Wang, L.; Chang, C.W.; Heinrich, S.; Dominguez, D.; Forgues, M.; Candia, J.; Hernandez, M.O.; Kelly, M.; Zhao, Y.; et al. Single-cell atlas of tumor clonal evolution in liver cancer. bioRxiv 2020. bioRxiv:2020.08.18.254748. [Google Scholar]
- Song, G.; Shi, Y.; Meng, L.; Ma, J.; Huang, S.; Zhang, J.; Wu, Y.; Li, J.; Lin, Y.; Yang, S.; et al. Single-cell transcriptomic analysis suggests two molecularly subtypes of intrahepatic cholangiocarcinoma. Nat. Commun. 2022, 13, 1642. [Google Scholar] [CrossRef]
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021, 39, 866–882.e11. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, C.; Zhang, Z.; Wu, H.; Li, X.; Zhang, Y.; Deng, N.; Dang, N.; Tang, G.; Yang, X.; et al. Cellular heterogeneity and transcriptomic profiles during intrahepatic cholangiocarcinoma initiation and progression. Hepatology 2022, 76, 1302–1317. [Google Scholar] [CrossRef]
- Ma, L.; Wang, L.; Khatib, S.A.; Chang, C.-W.; Heinrich, S.; Dominguez, D.A.; Forgues, M.; Candia, J.; Hernandez, M.O.; Kelly, M.; et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 75, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Fu, J.; Wang, X.; Lee, J.; Brar, G.; Escorcia, F.E.; Cam, M.; Xie, C. Characterization of Immunogenicity of Malignant Cells with Stemness in Intrahepatic Cholangiocarcinoma by Single-Cell RNA Sequencing. Stem Cells Int. 2022, 2022, 3558200. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, L.; Rayar, M.; Desille, M.; Turlin, B.; Fautrel, A.; Boucher, E.; Llamas-Gutierrez, F.; Meunier, B.; Boudjema, K.; Clément, B.; et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2013, 58, 1992–2000. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and Genetic Characterization of Cholangiocarcinoma Identifies Therapeutic Targets for Tyrosine Kinase Inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Overi, D.; Melandro, F.; Grimaldi, A.; Cardinale, V.; Di Matteo, S.; Mennini, G.; Rossi, M.; Alvaro, D.; Barnaba, V.; et al. Matrisome analysis of intrahepatic cholangiocarcinoma unveils a peculiar cancer-associated extracellular matrix structure. Clin. Proteom. 2019, 16, 37. [Google Scholar] [CrossRef] [Green Version]
- Fabris, L.; Andersen, J.B.; Fouassier, L. Intrahepatic cholangiocarcinoma: A single-cell resolution unraveling the complexity of the tumor microenvironment. J. Hepatol. 2020, 73, 1007–1009. [Google Scholar] [CrossRef]
- Kitano, Y.; Okabe, H.; Yamashita, Y.-I.; Nakagawa, S.; Saito, Y.; Umezaki, N.; Tsukamoto, M.; Yamao, T.; Yamamura, K.; Arima, K.; et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br. J. Cancer 2018, 118, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Stenzinger, A.; Andrulis, M.; Klauschen, F.; Joehrens, K.; Warth, A.; Renner, M.; Mehrabi, A.; et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br. J. Cancer 2013, 109, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xie, Y.-Q.; Gao, M.; Zhao, Y.; Franco, F.; Wenes, M.; Siddiqui, I.; Bevilacqua, A.; Wang, H.; Yang, H.; et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 2021, 22, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Moreau, J.M.; Velegraki, M.; Bolyard, C.; Rosenblum, M.D.; Li, Z. Transforming growth factor-β1 in regulatory T cell biology. Sci. Immunol. 2022, 7, eabi4613. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, E.; Winzer, R.; Rissiek, A.; Ricklefs, I.; Meyer-Schwesinger, C.; Ricklefs, F.L.; Bauche, A.; Behrends, J.; Reimer, R.; Brenna, S.; et al. CD73-mediated adenosine production by CD8 T cell-derived extracellular vesicles constitutes an intrinsic mechanism of immune suppression. Nat. Commun. 2021, 12, 5911. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Li, K.; Niu, N.; Shao, Y.; Ding, D.; Thomas, D.L.; Jing, H.; Fujiwara, K.; Hu, H.; Osipov, A.; et al. Immune cell atlas of cholangiocarcinomas reveals distinct tumor microenvironments and associated prognoses. J. Hematol. Oncol. 2022, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Moor, A.E.; Harnik, Y.; Ben-Moshe, S.; Massasa, E.E.; Rozenberg, M.; Eilam, R.; Halpern, K.B.; Itzkovitz, S. Spatial Reconstruction of Single Enterocytes Uncovers Broad Zonation along the Intestinal Villus Axis. Cell 2018, 175, 1156–1167.e15. [Google Scholar] [CrossRef] [Green Version]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Tóth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E.; et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542, 352–356. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Species | Tumor | Tissue | Tissue Dissociation | Cells | Sequenced Cells (N) | Annotated Cell Types | Summary | Ref. |
|---|---|---|---|---|---|---|---|---|
| Human | 9 HCC 10 iCCA | Needle biopsied | Tumor dissociation Kit (Miltenyi Biotech) | Unsorted single cell suspension | 349,107 | Malignant cells, CD4 T cells, CD8 T cells, B cells, CAFs, TAMs, TECs, HPC-like | Tumor transcriptomic diversity of HCC and iCCA is associated with patient outcome and T cell polarization; tumor-derived VEGF drives tumor microenvironment reprogramming. | [27] |
| Human | 5 iCCA 3 adjacent normal tissue | Surgical resected | Dispase (Roche), type IV collagenase (Sigma), DNase I (Roche) | Unsorted single cell suspension | 56,871 | Malignant cells, cholangiocytes, hepatocytes, CD4 T cells, CD8 T cells, B cells, NK cells, Macrophages, DCs, fibroblasts, endothelial cells | Malignant cells display remarkable inter-tumor heterogeneity; Tregs revealed highly immunosuppressive features. | [28] |
| Human | 2 iCCA | Surgical resected | Dispase (Roche), type IV collagenase (Sigma), DNase I (Roche) | EpCAM−CD45−CD31− cells | 13,150 | CAFs | Six subsets are defined; CD146+ vCAFs are the most dominant fibroblasts and interact with malignant cells via IL6/IL6R axis; tumor exosomal miR-9-5p enhances IL6 expression in vCAFs and contributes to iCCA progression through EZH2. | [28] |
| Human | 25 HCC 12 iCCA | Needle biopsied or surgical resection | Tumor dissociation Kit (Miltenyi Biotech) | Unsorted single cell suspension | 91,019 | Hepatocytes, cholangiocytes, CD4 T cells, CD8 T cells, B cells, CAFs, TAMs, TECs | Functional clonality of tumor cells could be a prognostic surrogate in liver cancer; tumor clonality is linked to polarized immune cell landscape and osteopontin is potential player in tumor cell evolution. | [29] |
| Human | 14 iCCA | Surgical resected | Tumor dissociation Kit (Miltenyi Biotech) | Unsorted single cell suspension | 144,878 | Malignant cells, CD4 T cells, CD8 T cells, B cells, NK cells, MAIT, macrophages, DCs, monocytes, fibroblasts, endothelial cells | Two subsets of iCCA are defined with distinct marker and immune microenvironment. | [30] |
| Mouse | 2 YAP/AKT iCCA | Collagenase D (Roche), Trypsin-EDTA (Gibco), DNase I (Roche) | Sorted Col1a1-GFP+ cells | 13,026 | CAFs | The majority of CAFs in iCCA are derived from HSC; iCAFs promote iCCA through HGF–MET axis; CAF-derived type I collagen contributes to stiffness but does not promote iCCA growth. | [31] | |
| Mouse | 1 YAP/AKT iCCA 1 KRAS/p19 iCCA | Collagenase D (Roche), Trypsin-EDTA (Gibco), DNase I (Roche) | Sorted Col1a1-GFP+ cells mixed with unsorted cell suspension | 11,836 | CAFs | |||
| Mouse | Notch/AKT | Liver Dissociation Kit (Miltenyi Biotech) | Unsorted single cell suspension | 51,897 | Malignant cells, cholangiocytes, T cells, B cells, NK cells, macrophages, DCs, fibroblasts, endothelial cells, neutrophils | Stress-responding subtype and proliferating subtype of iCCA tumor cells are identified. The interaction of fibroblasts and endothelial cells promote iCCA growth. | [32] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golino, J.L.; Wang, X.; Maeng, H.M.; Xie, C. Revealing the Heterogeneity of the Tumor Ecosystem of Cholangiocarcinoma through Single-Cell Transcriptomics. Cells 2023, 12, 862. https://doi.org/10.3390/cells12060862
Golino JL, Wang X, Maeng HM, Xie C. Revealing the Heterogeneity of the Tumor Ecosystem of Cholangiocarcinoma through Single-Cell Transcriptomics. Cells. 2023; 12(6):862. https://doi.org/10.3390/cells12060862
Chicago/Turabian StyleGolino, Jihye L., Xin Wang, Hoyoung M. Maeng, and Changqing Xie. 2023. "Revealing the Heterogeneity of the Tumor Ecosystem of Cholangiocarcinoma through Single-Cell Transcriptomics" Cells 12, no. 6: 862. https://doi.org/10.3390/cells12060862