
Recent Emerging Immunological Treatments for Primary Brain Tumors: Focus on Chemokine-Targeting Immunotherapies
 , , , , and
, , , , and
Abstract
:1. Introduction
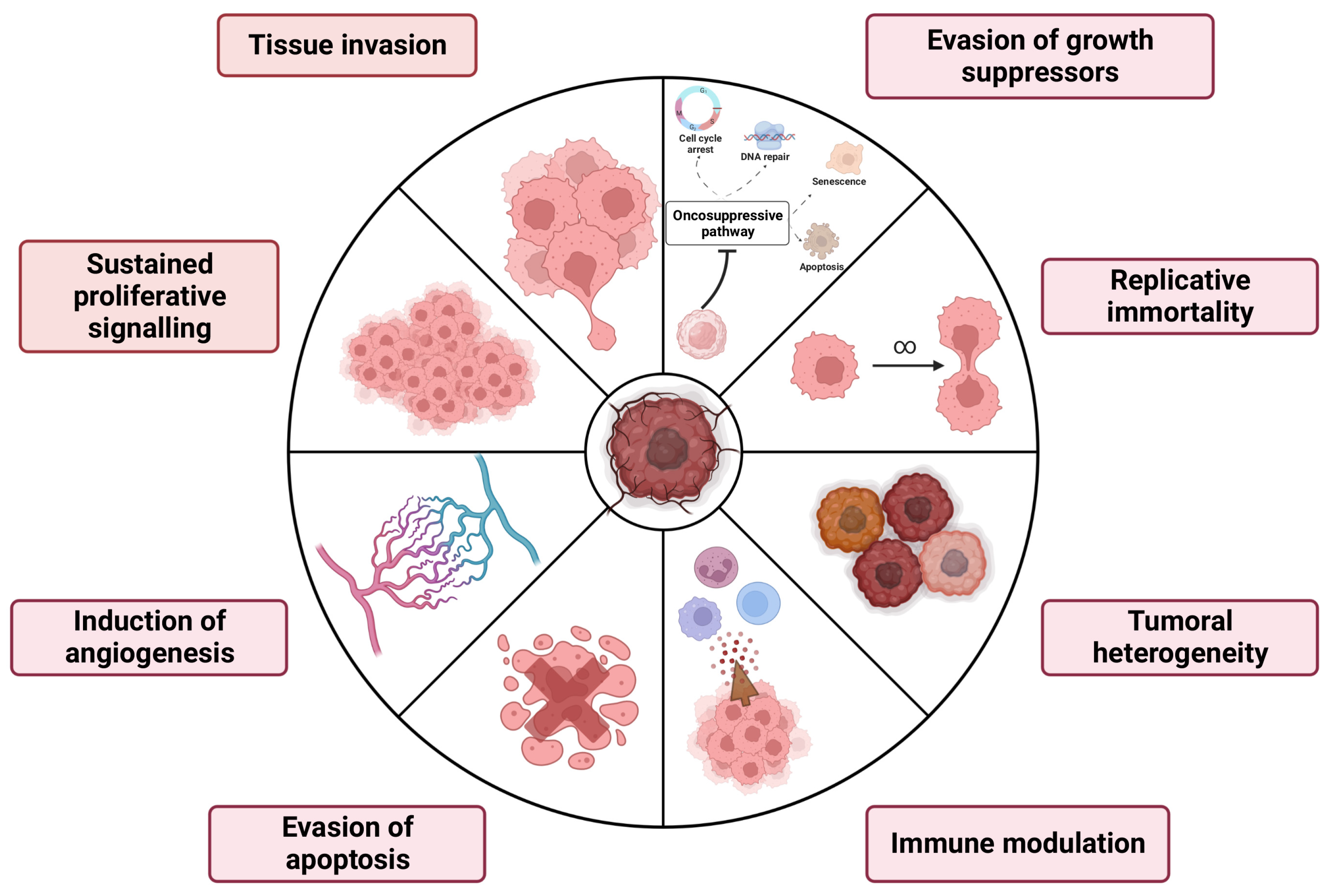
2. Primary Brain Tumors Hallmarks and Canonical Therapies
2.1. Brain Tumors: Classification
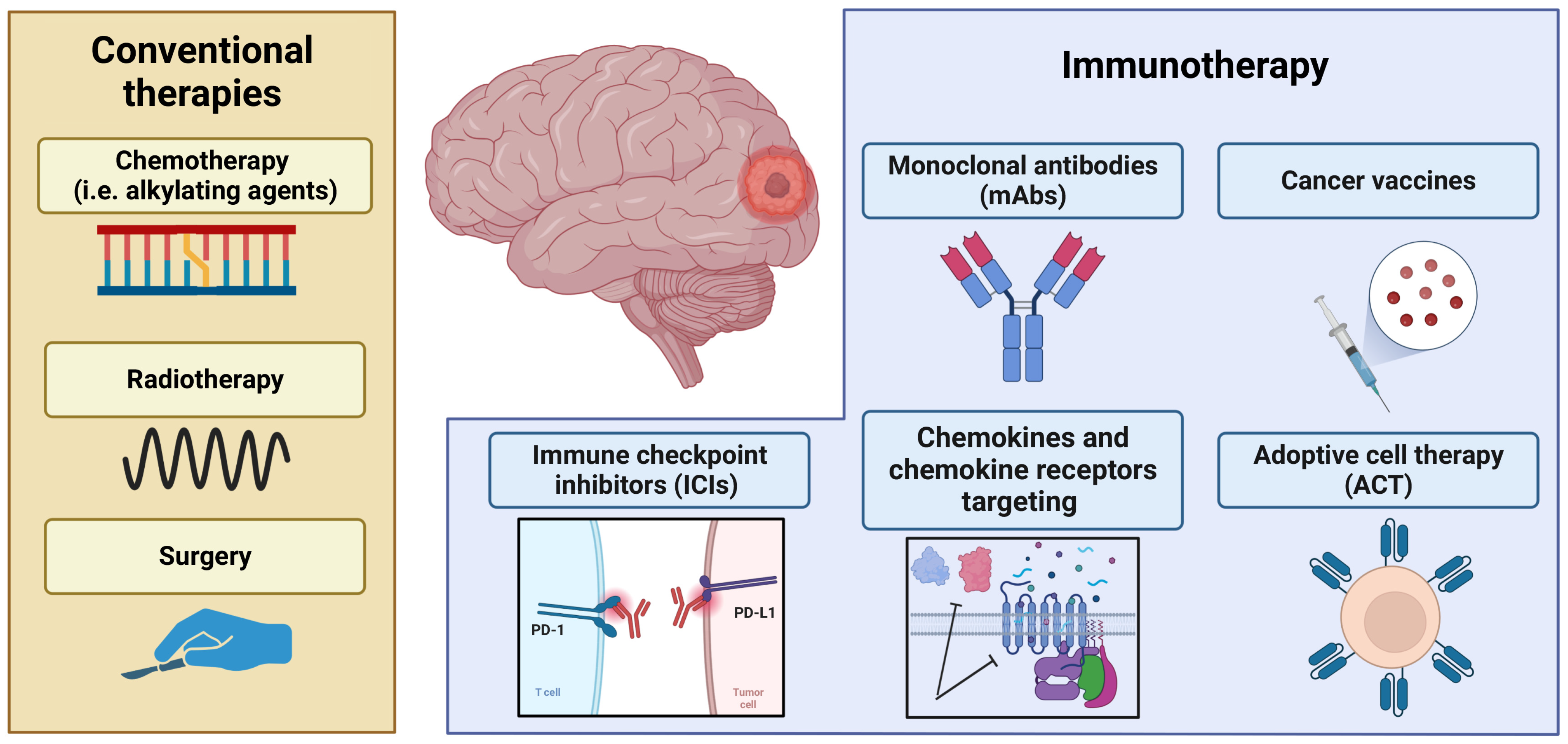
2.2. Brain Tumors: Current Clinical Approaches
3. Immunotherapeutic Approach to Control Primary Brain Cancers
3.1. Clinical Implication of Vaccines in Brain Cancers
3.2. Therapeutic Practice of Monoclonal Antibodies in Brain Cancers
3.3. Generation of Adoptive Cell Therapies to Counteract Brain Cancers
3.4. Immune Checkpoint Inhibitors
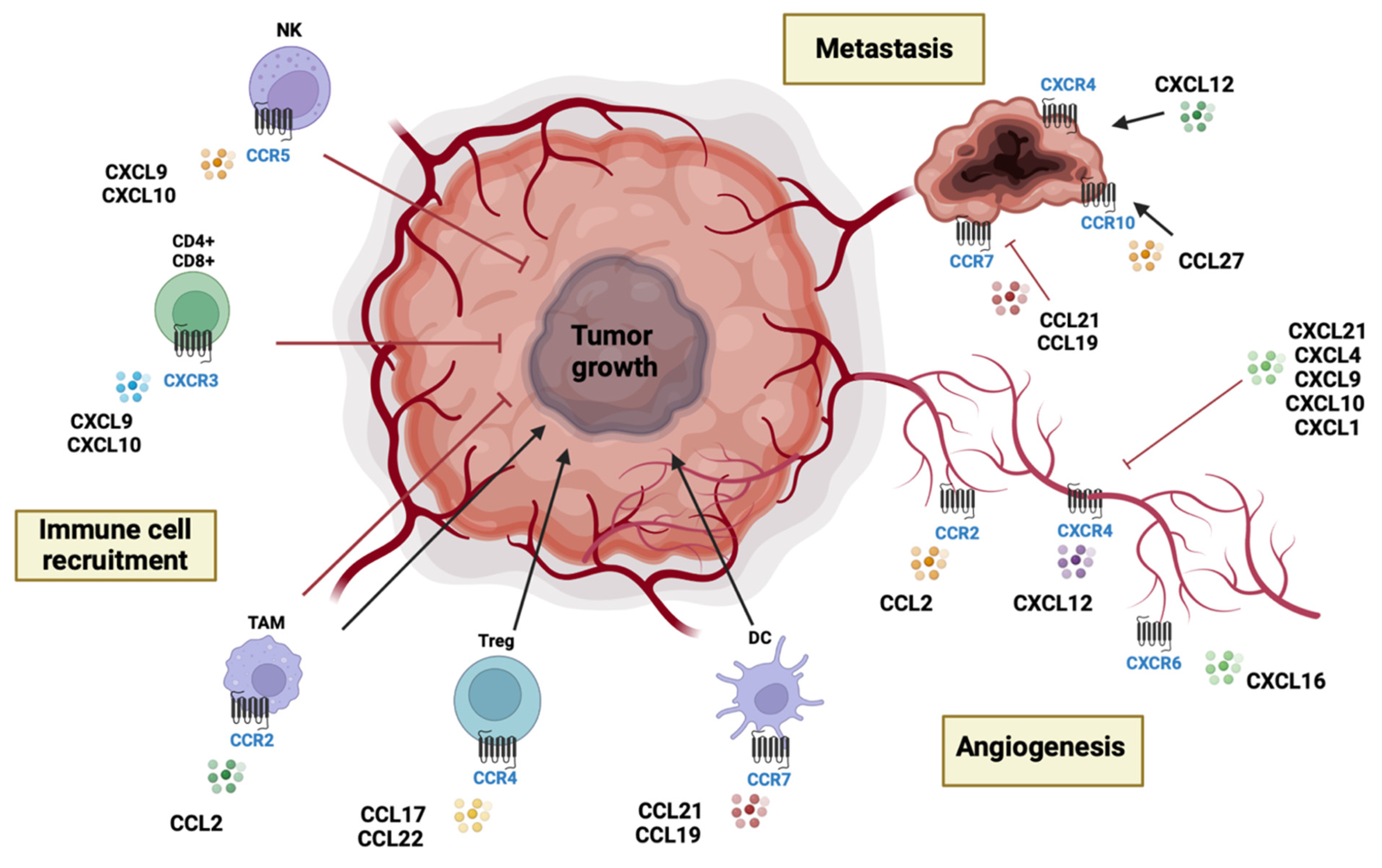
4. Chemokines and Chemokine Receptors in Brain Tumors
4.1. Chemokines and Chemokine Receptors: Classification and Biological Functions
4.2. CC Chemokines
4.3. CXC Chemokines
4.4. CX3C Chemokines
4.5. C Chemokines
5. Chemokine-Targeting Therapies in Primary Brain Cancers
5.1. Chemokine-Targeting Therapies: Preclinical Studies
5.2. Chemokine-Targeting Therapies: Clinical Studies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro. Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Chandana, S.R.; Movva, S.; Arora, M.; Singh, T. Primary brain tumors in adults. Am. Fam. Physician. 2008, 77, 1423. [Google Scholar]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.E.; Nibbs, R.J. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Chemokines and Chemokine Receptors as Therapeutic Targets in Inflammatory Bowel Disease; Pitfalls and Promise. J. Crohns. Colitis. 2018, 12, S641–S652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, S.L.; Godessart, N. Chemokines in autoimmunity: From pathology to therapeutics. Autoimmun. Rev. 2002, 1, 313–320. [Google Scholar] [CrossRef]
- Fallahi, P.; Ferrari, S.M.; Ragusa, F.; Ruffilli, I.; Elia, G.; Paparo, S.R.; Antonelli, A. Th1 chemokines in autoimmune endocrine disorders. J. Clin. Endocrinol. Metab. 2020, 105, 1046–1060. [Google Scholar] [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Geindreau, M.; Bruchard, M.; Vegran, F. Role of Cytokines and Chemokines in Angiogenesis in a Tumor Context. Cancers 2022, 14, 2446. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Casili, G.; Ardizzone, A.; Forte, S.; Colarossi, L.; Sava, S.; Paterniti, I.; Esposito, E.; Cuzzocrea, S.; Campolo, M. KYP-2047, an inhibitor of prolyl-oligopeptidase, reduces glioblastoma proliferation through angiogenesis and apoptosis modulation. Cancers 2021, 13, 3444. [Google Scholar] [CrossRef]
- Ardizzone, A.; Scuderi, S.A.; Giuffrida, D.; Colarossi, C.; Puglisi, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of Fibroblast Growth Factors Receptors (fgfrs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers 2020, 12, 3825. [Google Scholar] [CrossRef] [PubMed]
- Harter, P.N.; Braun, Y.; Plate, K.H. Classification of meningiomas-advances and controversies. Chin. Clin. Oncol. 2017, 6, S2. [Google Scholar] [CrossRef]
- Behling, F.; Hempel, J.-M.; Schittenhelm, J. Brain invasion in Meningioma—A prognostic potential worth exploring. Cancers 2021, 13, 3259. [Google Scholar] [CrossRef]
- Muhammad, S.; Niemelä, M. Management of oculomotor nerve schwannoma: Systematic review of literature and illustrative case. Surg. Neurol. Int. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocetti, E.; Trama, A.; Stiller, C.; Caldarella, A.; Soffietti, R.; Jaal, J.; Weber, D.C.; Ricardi, U.; Slowinski, J.; Brandes, A. Epidemiology of glial and non-glial brain tumours in Europe. Eur. J. Cancer 2012, 48, 1532–1542. [Google Scholar] [CrossRef]
- Kalasauskas, D.; Kosterhon, M.; Keric, N.; Korczynski, O.; Kronfeld, A.; Ringel, F.; Othman, A.; Brockmann, M.A. Beyond Glioma: The Utility of Radiomic Analysis for Non-Glial Intracranial Tumors. Cancers 2022, 14, 836. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Z.; Li, Z.; Wu, H.; Zheng, M.; Chang, Q.; Han, Y.; Cui, Z.; Liao, H.I.; Wang, T. Seldom-segment versus multi-segment intramedullary spinal cord gliomas: A comparative clinical study. Sci. China. Life Sci. 2018, 62, 862–864. [Google Scholar] [CrossRef]
- Sejda, A.; Grajkowska, W.; Trubicka, J.; Szutowicz, E.; Wojdacz, T.; Kloc, W.; Izycka-Swieszewska, E. WHO CNS5 2021 classification of gliomas: A practical review and road signs for diagnosing pathologists and proper patho-clinical and neuro-oncological cooperation. Folia. Neuropathol. 2022, 60, 137–152. [Google Scholar] [CrossRef]
- Farmanfarma, K.K.; Mohammadian, M.; Shahabinia, Z.; Hassanipour, S.; Salehiniya, H. Brain cancer in the world: An epidemiological review. World Cancer Res. J. 2019, 6, e1356. [Google Scholar]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.A. Cerebral edema and elevated intracranial pressure. CONTINUUM Lifelong Learn. Neurol. 2018, 24, 1588–1602. [Google Scholar] [CrossRef]
- Coomans, M.B.; van der Linden, S.D.; Gehring, K.; Taphoorn, M.J. Treatment of cognitive deficits in brain tumour patients: Current status and future directions. Curr. Opin. Oncol. 2019, 31, 540. [Google Scholar] [CrossRef] [PubMed]
- Alken, S.P.; D’Urso, P.; Saran, F.H. Managing teenage/young adult (TYA) brain tumors: A UK perspective. CNS Oncol. 2015, 4, 235–246. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal regulation of the blood–brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Langlands, J.M.; Kanekal, S.; Steino, A.; Mendez, A.; Karman, Z.; Toth, B.M.; Heredi-Szabo, K.; Brown, D. Dianhydrogalactitol (VAL-083) for the treatment of glioblastoma multiforme (GBM): Impact of glucose transporters for crossing the blood brain barrier (BBB). Cancer Res. 2022, 82, 1843. [Google Scholar] [CrossRef]
- Gabay, M.; Weizman, A.; Zeineh, N.; Kahana, M.; Obeid, F.; Allon, N.; Gavish, M. Liposomal carrier conjugated to APP-derived peptide for brain cancer treatment. Cell. Mol. Neurobiol. 2021, 41, 1019–1029. [Google Scholar] [CrossRef]
- Cano, A.; Espina, M.; Garcia, M.L. Recent advances on antitumor agents-loaded polymeric and lipid-based nanocarriers for the treatment of brain cancer. Curr. Pharm. Des. 2020, 26, 1316–1330. [Google Scholar] [CrossRef]
- Hettiarachchi, S.D.; Graham, R.M.; Mintz, K.J.; Zhou, Y.; Vanni, S.; Peng, Z.; Leblanc, R.M. Triple conjugated carbon dots as a nano-drug delivery model for glioblastoma brain tumors. Nanoscale 2019, 11, 6192–6205. [Google Scholar] [CrossRef] [PubMed]
- Hafazalla, K.; Sahgal, A.; Jaja, B.; Perry, J.R.; Das, S. Procarbazine, CCNU and vincristine (PCV) versus temozolomide chemotherapy for patients with low-grade glioma: A systematic review. Oncotarget 2018, 9, 33623. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Mehta, M.P. Low-grade glioma radiotherapy treatment and trials. Neurosurg. Clin. 2019, 30, 111–118. [Google Scholar] [CrossRef]
- Scaringi, C.; Agolli, L.; Minniti, G. Technical Advances in Radiation Therapy for Brain Tumors. Anticancer Res. 2018, 38, 6041–6045. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.M.; Lim, M.; Drake, C.G. Immunotherapy for Brain Cancer: Recent Progress and Future promiseimmunotherapy for Brain Cancer. Clin. Cancer Res. 2014, 20, 3651–3659. [Google Scholar] [CrossRef] [Green Version]
- Giotta Lucifero, A.; Luzzi, S.; Brambilla, I.; Trabatti, C.; Mosconi, M.; Savasta, S.; Foiadelli, T. Innovative therapies for malignant brain tumors: The road to a tailored cure. Acta Biomed. 2020, 91, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis. Treat. 2017, 3, 250–261. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Baxter, D. Active and passive immunization for cancer. Hum. Vaccin. Immunother. 2014, 10, 2123–2129. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Maus, M.V.; June, C.H. Immunotherapy for Brain Tumors. J. Clin. Oncol. 2017, 35, 2450–2456. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaugler, B.; Van den Eynde, B.; van der Bruggen, P.; Romero, P.; Gaforio, J.J.; De Plaen, E.; Lethe, B.; Brasseur, F.; Boon, T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 1994, 179, 921–930. [Google Scholar] [CrossRef]
- Lee, J.; Uy, B.R.; Liau, L.M. Brain Tumor Vaccines. Neurosurg. Clin. N. Am. 2021, 32, 225–234. [Google Scholar] [CrossRef]
- Elsamadicy, A.A.; Chongsathidkiet, P.; Desai, R.; Woroniecka, K.; Farber, S.H.; Fecci, P.E.; Sampson, J.H. Prospect of rindopepimut in the treatment of glioblastoma. Expert. Opin. Biol. Ther. 2017, 17, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Ochs, K.; Ott, M.; Bunse, T.; Sahm, F.; Bunse, L.; Deumelandt, K.; Sonner, J.K.; Keil, M.; von Deimling, A.; Wick, W.; et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology 2017, 6, e1328340. [Google Scholar] [CrossRef] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Boockvar, J.; Bodhinayake, I.; Brooks, C.; Chen, J.; Schwartz, J.; Rowinsky, E.; Reardon, D. It-02: Initiation of Clinical Studies with Sl-701, a Synthetic Multi-Peptide Vaccine with Enhanced Immunostimulatory Properties Targeting Multiple Glioma-Associated Antigens, in Adults with First Recurrence of Glioblastoma. Neuro. Oncol. 2014, 16, v110. [Google Scholar] [CrossRef] [Green Version]
- Buss, N.A.; Henderson, S.J.; Mcfarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharm. 2012, 12, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R. Rituximab (monoclonal anti-CD20 antibody): Mechanisms of action and resistance. Oncogene 2003, 22, 7359–7368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Liu, C.; Qi, H.; Zhou, J.; Wen, J.; Wu, D.; Xu, D.; Qin, M.; Ren, J.; Wang, Q.; et al. Systemic Delivery of Monoclonal Antibodies to the Central Nervous System for Brain Tumor Therapy. Adv. Mater. 2019, 31, e1805697. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Guo, C.C.; Chen, Z.P. Profile of nimotuzumab in the treatment of high-grade glioma. Onco. Targets 2015, 8, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Fecci, P.E.; Sampson, J.H. The current state of immunotherapy for gliomas: An eye toward the future. J. Neurosurg. 2019, 131, 657–666. [Google Scholar] [CrossRef]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef]
- Wachsmann, T.L.A.; Wouters, A.K.; Remst, D.F.G.; Hagedoorn, R.S.; Meeuwsen, M.H.; van Diest, E.; Leusen, J.; Kuball, J.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Comparing CAR and TCR engineered T cell performance as a function of tumor cell exposure. Oncoimmunology 2022, 11, 2033528. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused egfrviii-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Abril-Rodriguez, G.; Ribas, A. Snapshot: Immune Checkpoint Inhibitors. Cancer Cell 2017, 31, 848–848.e1. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Shitara, K.; Ajani, J.A.; Moehler, M.; Garrido, M.; Gallardo, C.; Shen, L.; Yamaguchi, K.; Wyrwicz, L.; Skoczylas, T.; Bragagnoli, A.C.; et al. Nivolumab plus chemotherapy or ipilimumab in gastro-oesophageal cancer. Nature 2022, 603, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Park, K.; Govindan, R.; Ready, N.; Reck, M.; Peters, S.; Dakhil, S.R.; Navarro, A.; Rodriguez-Cid, J.; Schenker, M.; et al. Nivolumab and Ipilimumab as Maintenance Therapy in Extensive-Disease Small-Cell Lung Cancer: Checkmate 451. J. Clin. Oncol. 2021, 39, 1349–1359. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Flechon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the checkmate 650 Trial. Cancer Cell 2020, 38, 489–499. [Google Scholar] [CrossRef]
- Chen, R.; Tao, Y.; Xu, X.; Shan, L.; Jiang, H.; Yin, Q.; Pei, L.; Cai, F.; Ma, L.; Yu, Y. The efficacy and safety of nivolumab, pembrolizumab, and atezolizumab in treatment of advanced non-small cell lung cancer. Discov. Med. 2018, 26, 155–166. [Google Scholar]
- Wong, J.S.L.; Kwok, G.G.W.; Tang, V.; Li, B.C.W.; Leung, R.; Chiu, J.; Ma, K.W.; She, W.H.; Tsang, J.; Lo, C.M.; et al. Ipilimumab and nivolumab/pembrolizumab in advanced hepatocellular carcinoma refractory to prior immune checkpoint inhibitors. J. Immunother. Cancer. 2021, 9, e001945. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.T.; Friedlander, T.W.; Hoimes, C.J.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma: Updated Results From a Phase 1/2 Open-label Study. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulovic, S.; Demey, W.; Ullen, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Marciscano, A.E.; Gulley, J.L. Avelumab demonstrates promise in advanced NSCLC. Oncotarget 2017, 8, 102767–102768. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, K.; Hubben, A.; Ahluwalia, M. The Role of Checkpoint Inhibitors in Glioblastoma. Target Oncol. 2019, 14, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Stanton, E.H.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. The role of chemokines in the pathophysiology of major depressive disorder. Int. J. Mol. Sci. 2019, 20, 2283. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Kojder, K.; Simińska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef]
- Darakhshan, S.; Hassanshahi, G.; Mofidifar, Z.; Soltani, B.; Karimabad, M.N. CXCL9/CXCL10 angiostasis CXC-chemokines in parallel with the CXCL12 as an angiogenesis CXC-chemokine are variously expressed in pre-eclamptic-women and their neonates. Pregnancy Hypertens. 2019, 17, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Gencer, S.; Evans, B.R.; van der Vorst, E.P.; Döring, Y.; Weber, C. Inflammatory chemokines in atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef] [PubMed]
- Henrot, P.; Prevel, R.; Berger, P.; Dupin, I. Chemokines in COPD: From implication to therapeutic use. Int. J. Mol. Sci. 2019, 20, 2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, I.; Dzierżęga-Lęcznar, A.; Stępień, K. Differential expression of inflammatory cytokines and chemokines in lipopolysaccharide-stimulated melanocytes from lightly and darkly pigmented skin. Exp. Dermatol. 2019, 28, 551–560. [Google Scholar] [CrossRef]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and chemokine receptors during COVID-19 infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Kimmel, R. Chemokines as drivers of the autoimmune destruction in type 1 diabetes: Opportunity for therapeutic intervention in consideration of an optimal treatment schedule. Front. Endocrinol. 2020, 11, 591083. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. Inflammatory cytokines and chemokines as therapeutic targets in heart failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. Regulation of the T cell response. Clin. Exp. Allergy 2006, 36, 1357–1366. [Google Scholar] [CrossRef]
- Zanganeh, E.; Soudi, S.; Hosseini, A.Z.; Khosrojerdi, A. Repeated intravenous injection of adipose tissue derived mesenchymal stem cells enhances Th1 immune responses in Leishmania major-infected BALB/c mice. Immunol. Lett. 2019, 216, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Matsuda, S.; Koyasu, S. Synergistic effects of IL-4 and IL-18 on IL-12-dependent IFN-γ production by dendritic cells. J. Immunol. 2000, 164, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Schindler, H.; Lutz, M.B.; Röllinghoff, M.; Bogdan, C. The production of IFN-γ by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J. Immunol. 2001, 166, 3075–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaylo-Moynihan, A.; Prizant, H.; Popović, M.; Fernandes, N.R.; Anderson, C.S.; Chiou, K.K.; Bell, H.; Schrock, D.C.; Schumacher, J.; Capece, T. Programming of distinct chemokine-dependent and-independent search strategies for Th1 and Th2 cells optimizes function at inflamed sites. Immunity 2019, 51, 298–309.E296. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A.; Mackay, C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today 1998, 19, 568–574. [Google Scholar] [CrossRef]
- Gauthier, M.; Kale, S.; Oriss, T.; Das, S.; Scholl, K.; Ray, P.; Wenzel, S.; Ray, A. A Dual Role of CXCR3 and CCR5 in T1 Inflammation in Severe Asthma. In TP9. TP009 Mechanistic and Translational Asthma Studies; American Thoracic Society: New York, NY, USA, 2021; p. A1409. [Google Scholar]
- Kumar, R.; Bhatia, M.; Pai, K. Role of Chemokines in the Pathogenesis of Visceral Leishmaniasis. Curr. Med. Chem. 2022, 29, 5441–5461. [Google Scholar] [CrossRef]
- Awane, M.; Andres, P.G.; Li, D.J.; Reinecker, H.-C. NF-κB-inducing kinase is a common mediator of IL-17-, TNF-α-, and IL-1β-induced chemokine promoter activation in intestinal epithelial cells. J. Immunol. 1999, 162, 5337–5344. [Google Scholar] [CrossRef]
- Jang, J.Y.; Choi, G.H.; Ji, S. IFN-γ or IL-4 polarization impacts the response of gingival fibroblasts to oral bacteria. J. Periodontal. Res. 2021, 56, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Nisar, S.; Maacha, S.; Carneiro-Lobo, T.C.; Akhtar, S.; Siveen, K.S.; Wani, N.A.; Rizwan, A.; Bagga, P.; Singh, M. Cytokine-chemokine network driven metastasis in esophageal cancer; promising avenue for targeted therapy. Mol. Cancer 2021, 20, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-stroma-inflammation networks promote pro-metastatic chemokines and aggressiveness characteristics in triple-negative breast cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [Green Version]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef] [PubMed]
- Palomino, D.C.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Grossman, J.G.; Nywening, T.M.; Belt, B.A.; Panni, R.Z.; Krasnick, B.A.; Denardo, D.G.; Hawkins, W.G.; Goedegebuure, S.P.; Linehan, D.C.; Fields, R.C. Recruitment of CCR2+ tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology 2018, 7, e1470729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonska, J.; Wu, C.F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, J.P.; Albrecht, J.D.; Alberti-Violetti, S.; Berti, E. CCR4 in cutaneous T-cell lymphoma: Therapeutic targeting of a pathogenic driver. Eur. J. Immunol. 2021, 51, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Chen, Y.-S.; Yao, Y.-D.; Chen, J.-Q.; Chen, J.-N.; Huang, S.-Y.; Zeng, Y.-J.; Yao, H.-R.; Zeng, S.-H.; Fu, Y.-S. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 2015, 6, 34758. [Google Scholar] [CrossRef] [Green Version]
- Strieter, R.M.; Burdick, M.D.; Gomperts, B.N.; Belperio, J.A.; Keane, M.P. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 593–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuesta-Mateos, C.; Juárez-Sánchez, R.; Mateu-Albero, T.; Loscertales, J.; Mol, W.; Terrón, F.; Muñoz-Calleja, C. Targeting cancer homing into the lymph node with a novel anti-CCR7 therapeutic antibody: The paradigm of CLL. Proc. Mabs 2021, 13, 1917484. [Google Scholar] [CrossRef]
- Sharma, I.; Singh, A.; Sharma, K.C.; Saxena, S. Gene expression profiling of chemokines and their receptors in low and high grade astrocytoma. Asian Pac. J. Cancer Prev. 2017, 18, 1307. [Google Scholar] [PubMed]
- Cho, H.R.; Kumari, N.; Thi Vu, H.; Kim, H.; Park, C.K.; Choi, S.H. Increased Antiangiogenic Effect by Blocking CCL2-dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study with Dynamic Susceptibility Contrast Perfusion MRI. Sci. Rep. 2019, 9, 11085. [Google Scholar] [CrossRef] [Green Version]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bajdak-Rusinek, K.; Kupnicka, P.; Kapczuk, P.; Simińska, D.; Chlubek, D.; Baranowska-Bosiacka, I. The role of CXCL16 in the pathogenesis of cancer and other diseases. Int. J. Mol. Sci. 2021, 22, 3490. [Google Scholar] [CrossRef]
- Yu, L.; Yang, X.; Xu, C.; Sun, J.; Fang, Z.; Pan, H.; Han, W. Comprehensive analysis of the expression and prognostic value of CXC chemokines in colorectal cancer. Int. Immunopharmacol. 2020, 89, 107077. [Google Scholar] [CrossRef]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their receptors: Multifaceted roles in cancer progression and potential value as cancer prognostic markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Chen, N.; Li, Y.; Zheng, H.; Lei, Q. CXCR6/CXCL16 functions as a regulator in metastasis and progression of cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2010, 1806, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Midavaine, É.; Côté, J.; Sarret, P. The multifaceted roles of the chemokines CCL2 and CXCL12 in osteophilic metastatic cancers. Cancer Metastasis Rev. 2021, 40, 427–445. [Google Scholar] [CrossRef]
- Shen, T.; Yang, Z.; Cheng, X.; Xiao, Y.; Yu, K.; Cai, X.; Xia, C.; Li, Y. CXCL8 induces epithelial-mesenchymal transition in colon cancer cells via the PI3K/Akt/NF-κB signaling pathway. Oncol. Rep. 2017, 37, 2095–2100. [Google Scholar] [CrossRef] [Green Version]
- Numan, Y.; Zhao, J.; Tang, S.; Zhang, Y.; Zhang, Q.; Jovanovic, B.; Vanderweele, D.J.; Morgans, A.K.; Cristofanilli, M.; Yu, J. Chemokine signaling and MAPK/ERK pathway for advanced prostate cancer treatment response. Am. Soc. Clin. Oncol. 2020, 38, TPS275. [Google Scholar] [CrossRef]
- Kremer, K.N.; Peterson, K.L.; Schneider, P.A.; Meng, X.W.; Dai, H.; Hess, A.D.; Smith, B.D.; Rodriguez-Ramirez, C.; Karp, J.E.; Kaufmann, S.H. CXCR4 chemokine receptor signaling induces apoptosis in acute myeloid leukemia cells via regulation of the Bcl-2 family members Bcl-XL, Noxa, and Bak. J. Biol. Chem. 2013, 288, 22899–22914. [Google Scholar] [CrossRef] [Green Version]
- Mortezaee, K. CXCL12/CXCR4 axis in the microenvironment of solid tumors: A critical mediator of metastasis. Life Sci. 2020, 249, 117534. [Google Scholar] [CrossRef]
- Terasaki, M.; Sugita, Y.; Arakawa, F.; Okada, Y.; Ohshima, K.; Shigemori, M. CXCL12/CXCR4 signaling in malignant brain tumors: A potential pharmacological therapeutic target. Brain Tumor Pathol. 2011, 28, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.H.; Mentlein, R.; Knerlich, F.; Kruse, M.L.; Mehdorn, H.M.; Held-Feindt, J. Expression of stem cell markers in human astrocytomas of different WHO grades. J. Neurooncol. 2008, 86, 31–45. [Google Scholar] [CrossRef]
- Korbecki, J.; Siminska, D.; Kojder, K.; Grochans, S.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Fractalkine/CX3CL1 in Neoplastic Processes. Int. J. Mol. Sci. 2020, 21, 3723. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski-Lenoir, D.; Chen, S.; Feng, L.; Maki, R.; Bacon, K.B. Characterization of fractalkine in rat brain cells: Migratory and activation signals for CX3CR-1-expressing microglia. J. Immunol. 1999, 163, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Sciume, G.; Soriani, A.; Piccoli, M.; Frati, L.; Santoni, A.; Bernardini, G. CX3CR1/CX3CL1 axis negatively controls glioma cell invasion and is modulated by transforming growth factor-beta1. Neuro. Oncol. 2010, 12, 701–710. [Google Scholar] [CrossRef]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Bird, E.V.; Iannitti, T.; Christmas, C.R.; Obara, I.; Andreev, V.I.; King, A.E.; Boissonade, F.M. A Novel Role for Lymphotactin (XCL1) Signaling in the Nervous System: XCL1 Acts via its Receptor XCR1 to Increase Trigeminal Neuronal Excitability. Neuroscience 2018, 379, 334–349. [Google Scholar] [CrossRef] [PubMed]
- Cairns, C.M.; Gordon, J.R.; Li, F.; Baca-Estrada, M.E.; Moyana, T.; Xiang, J. Lymphotactin expression by engineered myeloma cells drives tumor regression: Mediation by CD4+ and CD8+ T cells and neutrophils expressing XCR1 receptor. J. Immunol. 2001, 167, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; e Sousa, C.R. NK cells stimulate recruitment of cdc1 into the tumor microenvironment promoting cancer immune control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [Green Version]
- Sehedic, D.; Chourpa, I.; Tetaud, C.; Griveau, A.; Loussouarn, C.; Avril, S.; Legendre, C.; Lepareur, N.; Wion, D.; Hindre, F.; et al. Locoregional Confinement and Major Clinical Benefit of (188)Re-Loaded CXCR4-Targeted Nanocarriers in an Orthotopic Human to Mouse Model of Glioblastoma. Theranostics 2017, 7, 4517–4536. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.; Shubina, A.; Sokolov, D.; Selkov, S.; Baranov, V.; Kiselev, A. CXCR4-targeted modular peptide carriers for efficient anti-VEGF sirna delivery. Int. J. Pharm. 2016, 515, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Alsulami, M.; Arbab, S.A.; Ara, R.; Shankar, A.; Iskander, A.; Angara, K.; Jain, M.; Bagher-Ebadian, H.; Achyut, B.R.; et al. Targeting Bone Marrow to Potentiate the Anti-Tumor Effect of Tyrosine Kinase Inhibitor in Preclinical Rat Model of Human Glioblastoma. Int. J. Cancer Res. 2016, 12, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; La Pietra, V.; Piccarducci, R.; Pietrobono, D.; Cavallini, C.; D’Amore, V.M.; Cerofolini, L.; Giuntini, S.; Russomanno, P.; Puxeddu, M.; et al. CXCR4 antagonism sensitizes cancer cells to novel indole-based MDM2/4 inhibitors in glioblastoma multiforme. Eur. J. Pharm. 2021, 897, 173936. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neurooncol. 2019, 143, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Wang, B.; Chen, Y.; Liu, H.; Shi, L. Novel CXCR4 Inhibitor CPZ1344 Inhibits the Proliferation, Migration and Angiogenesis of Glioblastoma. Pathol. Oncol. Res. 2020, 26, 2597–2604. [Google Scholar] [CrossRef]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. 2016, 35, 55. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Vitale, F.; Vetuschi, A.; Pompili, S.; Rossi, G.; Marampon, F.; Richardson, P.J.; Patient, L.; et al. The novel CXCR4 antagonist, PRX177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models. Tumour. Biol. 2017, 39, 1010428317695528. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Hu, Y.; Tang, L.; Yin, S.; Lv, L.; Zhou, P. Targeting CXCR4 to suppress glioma-initiating cells and chemoresistance in glioma. Cell Biol. Int. 2022, 46, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wu, H.; Wang, H.; Li, Y.; Zhang, L.; Zhu, L.; Wang, W.; Zhou, J.; Fu, Y.; Chen, S.; et al. N-terminal polypeptide derived from vmip-II exerts its antitumor activity by inhibiting the CXCR4 pathway in human glioma. Int. J. Oncol. 2017, 50, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Gagner, J.P.; Sarfraz, Y.; Ortenzi, V.; Alotaibi, F.M.; Chiriboga, L.A.; Tayyib, A.T.; Douglas, G.J.; Chevalier, E.; Romagnoli, B.; Tuffin, G.; et al. Multifaceted C-X-C Chemokine Receptor 4 (CXCR4) Inhibition Interferes with Anti-Vascular Endothelial Growth Factor Therapy-Induced Glioma Dissemination. Am. J. Pathol. 2017, 187, 2080–2094. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yang, B. Double-Targeted Knockdown of mir-21 and CXCR4 Inhibits Malignant Glioma Progression by Suppression of the PI3K/AKT and Raf/MEK/ERK Pathways. Biomed. Res. Int. 2020, 2020, 7930160. [Google Scholar] [CrossRef]
- Ward, S.A.; Warrington, N.M.; Taylor, S.; Kfoury, N.; Luo, J.; Rubin, J.B. Reprogramming Medulloblastoma-Propagating Cells by a Combined Antagonism of Sonic Hedgehog and CXCR4. Cancer Res. 2017, 77, 1416–1426. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Abraham, M.; Bulvik, B.; Dery, E.; Weiss, I.D.; Barashi, N.; Abramovitch, R.; Wald, H.; Harel, Y.; Olam, D.; et al. CXCR4 Promotes Neuroblastoma Growth and Therapeutic Resistance through mir-15a/16-1-Mediated ERK and BCL2/Cyclin D1 Pathways. Cancer Res. 2018, 78, 1471–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Gascon, S.; Giraldo Solano, A.; El Kheir, W.; Therriault, H.; Berthelin, P.; Cattier, B.; Marcos, B.; Virgilio, N.; Paquette, B.; Faucheux, N.; et al. Characterization and Mathematical Modeling of Alginate/Chitosan-Based Nanoparticles Releasing the Chemokine CXCL12 to Attract Glioblastoma Cells. Pharmaceutics 2020, 12, 356. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Stafford, J.H.; Liu, S.C.; Chernikova, S.B.; Merchant, M.; Recht, L.; Martin Brown, J. SDF-1 Blockade Enhances Anti-VEGF Therapy of Glioblastoma and Can Be Monitored by MRI. Neoplasia 2017, 19, 1–7. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Z.; Wu, R.; Yao, Q.; Gu, Z.; Liu, M. Correlation of C-X-C chemokine receptor 2 upregulation with poor prognosis and recurrence in human glioma. Onco. Targets Ther. 2015, 8, 3203–3209. [Google Scholar] [CrossRef] [Green Version]
- Angara, K.; Borin, T.F.; Rashid, M.H.; Lebedyeva, I.; Ara, R.; Lin, P.C.; Iskander, A.; Bollag, R.J.; Achyut, B.R.; Arbab, A.S. CXCR2-Expressing Tumor Cells Drive Vascular Mimicry in Antiangiogenic Therapy-Resistant Glioblastoma. Neoplasia 2018, 20, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Acker, G.; Zollfrank, J.; Jelgersma, C.; Nieminen-Kelha, M.; Kremenetskaia, I.; Mueller, S.; Ghori, A.; Vajkoczy, P.; Brandenburg, S. The CXCR2/CXCL2 signalling pathway—An alternative therapeutic approach in high-grade glioma. Eur. J. Cancer 2020, 126, 106–115. [Google Scholar] [CrossRef]
- Urbantat, R.M.; Jelgersma, C.; Vajkoczy, P.; Brandenburg, S.; Acker, G. Combining TMZ and SB225002 induces changes of CXCR2 and VEGFR signalling in primary human endothelial cells in vitro. Oncol. Rep. 2022, 48, 158. [Google Scholar] [CrossRef]
- Dery, L.; Charest, G.; Guerin, B.; Akbari, M.; Fortin, D. Chemoattraction of Neoplastic Glial Cells with CXCL10, CCL2 and CCL11 as a Paradigm for a Promising Therapeutic Approach for Primary Brain Tumors. Int. J. Mol. Sci. 2021, 22, 2150. [Google Scholar] [CrossRef]
- Yu-Ju Wu, C.; Chen, C.H.; Lin, C.Y.; Feng, L.Y.; Lin, Y.C.; Wei, K.C.; Huang, C.Y.; Fang, J.Y.; Chen, P.Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro Oncol. 2020, 22, 253–266. [Google Scholar] [CrossRef]
- Lu, B.; Zhou, Y.; Su, Z.; Yan, A.; Ding, P. Effect of CCL2 sirna on proliferation and apoptosis in the U251 human glioma cell line. Mol. Med. Rep. 2017, 16, 3387–3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudati, E.; Curro, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef]
- Salazar, N.; Carlson, J.C.; Huang, K.; Zheng, Y.; Oderup, C.; Gross, J.; Jang, A.D.; Burke, T.M.; Lewen, S.; Scholz, A.; et al. A Chimeric Antibody against ACKR3/CXCR7 in Combination with TMZ Activates Immune Responses and Extends Survival in Mouse GBM Models. Mol. Ther. 2018, 26, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhao, Q.; Kong, L.Y.; Wang, J.; Yan, J.; Xia, X.; Jia, Z.; Heimberger, A.B.; Li, S. Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Sci. Adv. 2021, 7, eabc2511. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 2022, 31, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.; Hsu, S.H.; Blanco, A.; Buryanek, J.; Day, A.L.; Mcguire, M.F.; Brown, R.E. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience 2016, 3, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Urbantat, R.M.; Blank, A.; Kremenetskaia, I.; Vajkoczy, P.; Acker, G.; Brandenburg, S. The CXCL2/IL8/CXCR2 Pathway Is Relevant for Brain Tumor Malignancy and Endothelial Cell Function. Int. J. Mol. Sci. 2021, 22, 2634. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; lafrankie, D.; et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin. Cancer Res. 2018, 24, 4643–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.P.; Nagpal, S.; Iv, M.; Soltys, S.G.; Bertrand, S.; Pelpola, J.S.; Ball, R.; Yang, J.; Sundaram, V.; Lavezo, J.; et al. Macrophage Exclusion after Radiation Therapy (MERT): A First in Human Phase I/II Trial using a CXCR4 Inhibitor in Glioblastoma. Clin. Cancer Res. 2019, 25, 6948–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| First Author | Year | Molecular Target | Analysis/Outcome | References |
|---|---|---|---|---|
| Séhédic D. | 2017 | CXCR4 | The study analyzed the efficacy of rhenium-loaded nanocapsules expressing on their surface an anti-CXCR4, function-blocking antibody (12G5-LNC188Re) in an orthotopic in vivo GBM model. | [132] |
| Shaaban S. | 2016 | CXCR4 | The study evaluated whether whole body irradiation (WBIR) or a CXCR4 antagonist (AMD3100) potentiates the efficacy of vatalanib in an orthotopic in vivo GBM model. | [134] |
| Daniele S. | 2021 | CXCR4 | The in vitro study evaluated whether CXCR4 inhibition enhances the sensitivity of glioma cells to MDM2/4 inhibitors. | [135] |
| Wu A. | 2019 | CXCR4 | The study evaluated combination therapy with anti-CXCR4 and anti-PD-1 therapeutic antibodies in an in vivo murine glioma model. | [136] |
| Luo Z. | 2020 | CXCR4 | The study evaluated the efficacy of a novel CXCR4 inhibitor in a in vitro GBM model. | [137] |
| Mercurio L. | 2016 | CXCR4 | The study evaluated the effects of a novel CXCR4 antagonist (Peptide R) against glioblastoma in vitro and in vivo. | [138] |
| Gravina G.L. | 2017 | CXCR4 | The effects of a CXCR4 inhibitor (PRX177561) were evaluated in vitro, on several different glioblastoma cell lines, and in vivo, using a murine xenograft model. | [139] |
| Gravina G.L. | 2017 | CXCR4 | The study evaluated the efficacy of combination therapy with bevacizumab, sunitinib, and an anti-CXCR4 molecule (PRX177561) by using in vitro models as well as in vivo xenograft murine models. | [140] |
| Yang Q. | 2017 | CXCR4 | The in vitro study evaluated the effects of a novel designed CXCR4-inhibiting peptide called NT21MP, derived from vMIP-II, on glioma cell lines. | [142] |
| Liu F. | 2020 | CXCR4 | The study analyzed the effects of the inhibition of miR-21 and/or CXCR4 in vitro on glioma cells and in vivo in murine xenograft glioma models. | [144] |
| Ward S.A. | 2017 | CXCR4 | The study evaluated the therapeutic potential of dual CXCR4 and SHH inhibition in the treatment of medulloblastoma in an in vivo murine model. | [145] |
| Klein S. | 2017 | CXCR4 | The study analyzed the role of CXCR4 in neuroblastoma growth and the therapeutic potential of CXCR4 inhibition in neuroblastoma treatment both in vitro and in vivo. | [146] |
| Gascon S. | 2020 | CXCR4 | The study explored a novel therapeutic strategy for GBM treatment, using non-toxic CXCL12-loaded alginate/chitosan-based nanoparticles. The effects and toxicity of nanoparticles were tested in vitro. | [148] |
| Deng L. | 2017 | CXCL12 | This study evaluated in an orthotopic in vivo model whether OLA-PEG or NOX-A12 enhanced the antitumor effects of anti–VEGF therapeutic agents. | [149] |
| Acker G. | 2019 | CXCR2 | The study investigated the role of the CXCR2 pathway in glioma biology and the therapeutic potential of its inhibition using both in vitro and in vivo models. | [152] |
| Urbantat R.M. | 2022 | CXCR2 | The in vitro study analyzed the proangiogenic pathways following combined treatment with temozolomide and SB225002, a CXCR2 inhibitor, using primary endothelial cells which mimicked the GBM tumor microenvironment. | [153] |
| Dery L. | 2021 | CXCL10, CCL2 and CCL11 | In vitro and in vivo evaluation of three chemoattractants, CXCL10, CCL2, and CCL11, released by a biodegradable hydrogel (GlioGel) to produce a migration of tumor cells toward a therapeutic trap. | [154] |
| Yu-Ju Wu C. | 2020 | CCL5 | Using an in vitro model, this study investigated the mechanisms by which CCL5 facilitates the migratory and invasive activity of human glioma cells. | [155] |
| Lu B. | 2017 | CCL2 | The study evaluated the effects of transfection with a CCL2 siRNA into a human glioma cell line. | [156] |
| Cho H.R. | 2019 | CCL2 | Using a CCL2 inhibitor in an in vivo murine model, the potential value of CCL2 inhibition in combination with anti-VEGF agents in GBM was studied. | [112] |
| Laudati E. | 2017 | CCR5 | The authors investigated in vitro the effects of a CCR5 receptor blockade on microglia-glioma interaction through the use of maraviroc, a CCR5 blocker. | [157] |
| Salazar N. | 2018 | CXCR7 | The study analyzed the safety and efficacy of a single chain FV-human FC-immunoglobulin G1 antibody, X7A, to target ACKR3 in in vivo and in vitro GBM models. | [158] |
| Flores-Toro J.A. | 2020 | CCR2 | This study evaluated the combination of a PD-1 blockade and CCR2 inhibition in anti-PD-1-resistant gliomas using an in vivo murine model. | [113] |
| Wang G. | 2022 | CXCL11 | This in vivo and in vitro study investigated the activity of an oncolytic adenovirus (oAds) expressing the chemokine CXCL11 on the infiltration of CAR-T-cells and the reprogramming of the immunosuppressive tumor microenvironment. | [160] |
| First Author | Year | Molecular Target | Analysis/Outcome | References |
|---|---|---|---|---|
| Thomas R, P. | 2019 | CXCR4 | Phase I/II study to evaluate the safety and efficacy of “Macrophage Exclusion after Radiation Therapy”, using plerixafor, in glioblastoma patients. | [164] |
| Lee E, Q. | 2018 | CXCR4 | Phase I clinical study to determine the safety of the combination therapy of bevacizumab and plerixafor in patients with high-grade glioma. | [163] |
| Rios A. | 2016 | CXCR4 | Case of a patient (66 y.o. male) treated after standard chemo-radiotherapy with combination therapy including a CXCR4 inhibitor, plerixafor, showing a significant clinical response. | [161] |
| Urbantat R, M. | 2021 | CXCL2/IL-8 | Evaluation of CXCL2 and IL-8 expression in GBM patients’ brain tissue. Analysis of correlations between the gene expression of proangiogenic factors and patients’ overall survival | [162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardizzone, A.; Basilotta, R.; Filippone, A.; Crupi, L.; Lanza, M.; Lombardo, S.P.; Colarossi, C.; Sciacca, D.; Cuzzocrea, S.; Esposito, E.; et al. Recent Emerging Immunological Treatments for Primary Brain Tumors: Focus on Chemokine-Targeting Immunotherapies. Cells 2023, 12, 841. https://doi.org/10.3390/cells12060841
Ardizzone A, Basilotta R, Filippone A, Crupi L, Lanza M, Lombardo SP, Colarossi C, Sciacca D, Cuzzocrea S, Esposito E, et al. Recent Emerging Immunological Treatments for Primary Brain Tumors: Focus on Chemokine-Targeting Immunotherapies. Cells. 2023; 12(6):841. https://doi.org/10.3390/cells12060841
Chicago/Turabian StyleArdizzone, Alessio, Rossella Basilotta, Alessia Filippone, Lelio Crupi, Marika Lanza, Sofia Paola Lombardo, Cristina Colarossi, Dorotea Sciacca, Salvatore Cuzzocrea, Emanuela Esposito, and et al. 2023. "Recent Emerging Immunological Treatments for Primary Brain Tumors: Focus on Chemokine-Targeting Immunotherapies" Cells 12, no. 6: 841. https://doi.org/10.3390/cells12060841