

Increased Radiation Sensitivity in Patients with Phelan-McDermid Syndrome
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
3. Results
3.1. A Case of a Phelan-McDermid Syndrome Patient with an Atypical Teratoid/Rhabdoid Tumor
3.2. Individual Radiation Sensitivity of 20 Patients with Phelan-McDermid Syndrome
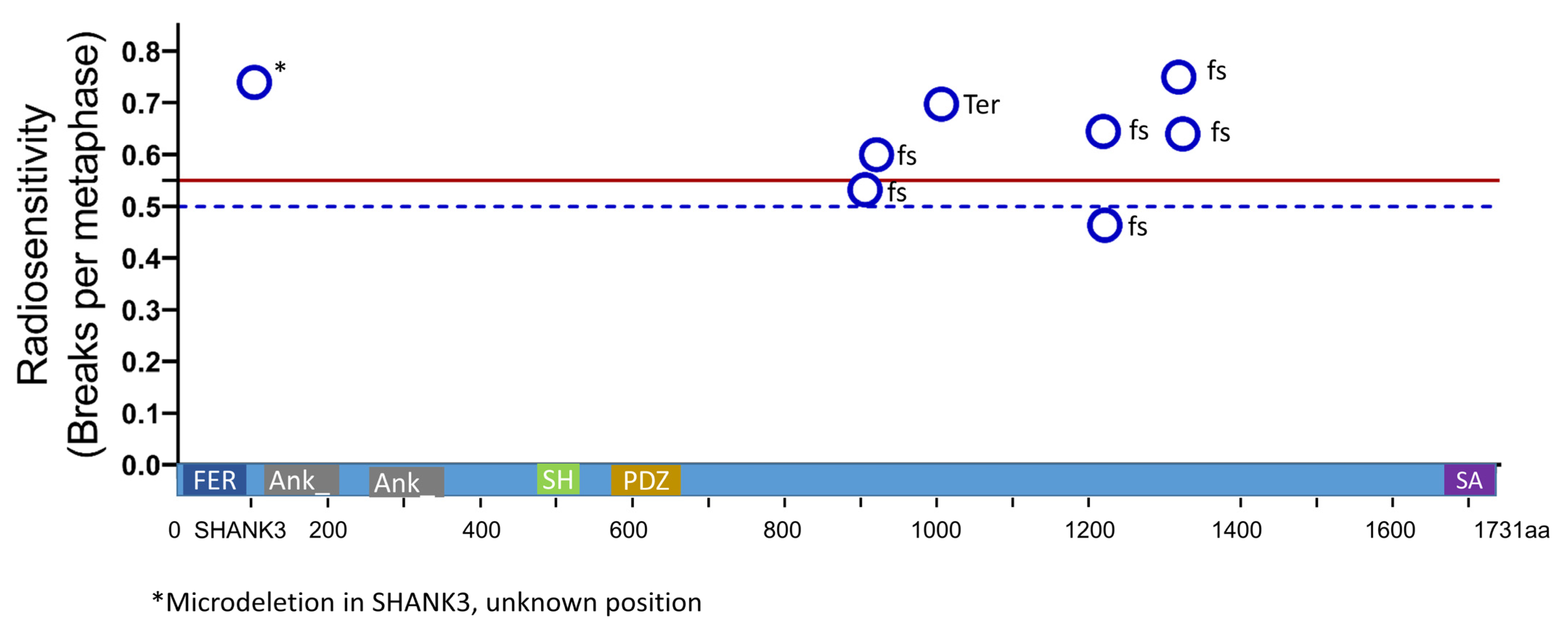
3.3. Mutation Type and Radiation Sensitivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, L.; Yao, Z.Y.; Wu, X.; He, S.R.; Liu, Y.M.; Wang, X.Y.; Cao, D.Z.; Yang, X.K.; Zhao, J.B.; Ren, Z.; et al. Phelan-McDermid Syndrome in Pediatric Patients With Novel Mutations: Genetic and Phenotypic Analyses. Front. Pediatr. 2022, 10, 888001. [Google Scholar] [CrossRef]
- Posserud, M.; Lundervold, A.J.; Lie, S.A.; Gillberg, C. The prevalence of autism spectrum disorders: Impact of diagnostic instrument and non-response bias. Soc. Psychiatry Psychiatr. Epidemiol. 2010, 45, 319–327. [Google Scholar] [CrossRef]
- Frazier, T.W.; Thompson, L.; Youngstrom, E.A.; Law, P.; Hardan, A.Y.; Eng, C.; Morris, N. A twin study of heritable and shared environmental contributions to autism. J. Autism Dev. Disord. 2014, 44, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancur, C.; Buxbaum, J.D. SHANK3 haploinsufficiency: A “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol. Autism 2013, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, K.; Rogers, R.C.; Boccuto, L. Phelan-McDermid Syndrome. In GeneReviews((R)); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.D.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkel, L.C.; Aref-Eshghi, E.; Rooney, K.; Kerkhof, J.; Levy, M.A.; McConkey, H.; Rogers, R.C.; Phelan, K.; Sarasua, S.M.; Jain, L.; et al. DNA methylation epi-signature is associated with two molecularly and phenotypically distinct clinical subtypes of Phelan-McDermid syndrome. Clin. Epigenet. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Tabet, A.C.; Rolland, T.; Ducloy, M.; Levy, J.; Buratti, J.; Mathieu, A.; Haye, D.; Perrin, L.; Dupont, C.; Passemard, S.; et al. A framework to identify contributing genes in patients with Phelan-McDermid syndrome. NPJ Genom Med. 2017, 2, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, K.; Boccuto, L.; Powell, C.M.; Boeckers, T.M.; van Ravenswaaij-Arts, C.; Rogers, R.C.; Sala, C.; Verpelli, C.; Thurm, A.; Bennett, W.E., Jr.; et al. Phelan-McDermid syndrome: A classification system after 30 years of experience. Orphanet J. Rare Dis. 2022, 17, 27. [Google Scholar] [CrossRef]
- Douglas, P.; Zhong, J.; Ye, R.; Moorhead, G.B.; Xu, X.; Lees-Miller, S.P. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol. Cell. Biol. 2010, 30, 1368–1381. [Google Scholar] [CrossRef] [Green Version]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Arakawa, Y.; Terada, Y.; Takeuchi, Y.; Mineharu, Y.; Sumiyoshi, S.; Tokunaga, S.; Nakajima, K.; Kawabata, N.; Tanaka, K.; et al. Whole-genome sequencing analysis of an atypical teratoid/rhabdoid tumor in a patient with Phelan-McDermid syndrome: A case report and systematic review. Brain Tumor Pathol. 2022, 39, 232–239. [Google Scholar] [CrossRef]
- Rajaraman, P.; Hauptmann, M.; Bouffler, S.; Wojcik, A. Human individual radiation sensitivity and prospects for prediction. Ann. ICRP 2018, 47, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Distel, L.V.; Neubauer, S.; Keller, U.; Sprung, C.N.; Sauer, R.; Grabenbauer, G.G. Individual differences in chromosomal aberrations after in vitro irradiation of cells from healthy individuals, cancer and cancer susceptibility syndrome patients. Radiother. Oncol. 2006, 81, 257–263. [Google Scholar] [CrossRef]
- Sharma, R.; Lewis, S.; Wlodarski, M.W. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front. Pediatr. 2020, 8, 570084. [Google Scholar] [CrossRef] [PubMed]
- Scott, D. Chromosomal radiosensitivity, cancer predisposition and response to radiotherapy. Strahlenther. Onkol. 2000, 176, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Auer, J.; Keller, U.; Schmidt, M.; Ott, O.; Fietkau, R.; Distel, L.V. Individual radiosensitivity in a breast cancer collective is changed with the patients’ age. Radiol. Oncol. 2014, 48, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, B.; Ellmann, A.; Mayo, T.; Auer, J.; Haas, M.; Hecht, M.; Fietkau, R.; Distel, L.V. Rate of individuals with clearly increased radiosensitivity rise with age both in healthy individuals and in cancer patients. BMC Geriatr. 2018, 18, 105. [Google Scholar] [CrossRef]
- Savage, J.R.; Simpson, P. On the scoring of FISH-"painted" chromosome-type exchange aberrations. Mutat. Res. 1994, 307, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Volpe, S.; Bondiau, P.Y.; Claude, L.; Claren, A.; Padovani, L.; AlGhamdi, H.; Duhil De Benaze, G.; Opitz, L.; Baudin, G.; Dejean, C.; et al. Postsurgical geometrical variations of tumor bed and brainstem during photon and proton therapy for pediatric tumors of the posterior fossa: Dosimetric impact and predictive factors. Strahlenther. Onkol. 2021, 197, 1113–1123. [Google Scholar] [CrossRef]
- Bernhardt, D.; Konig, L.; Grosu, A.; Wiestler, B.; Rieken, S.; Wick, W.; Gempt, J.; Krieg, S.M.; Schmidt-Graf, F.; Sahm, F.; et al. DEGRO practical guideline for central nervous system radiation necrosis part 1: Classification and a multistep approach for diagnosis. Strahlenther. Onkol. 2022, 198, 873–883. [Google Scholar] [CrossRef]
- Elsayad, K.; Kriz, J.; Samhouri, L.; Haverkamp, U.; Straeter, R.; Stummer, W.; Eich, H.T. Long-term survival following additive radiotherapy in patients with atypical teratoid rhabdoid tumors. Strahlenther. Onkol. 2016, 192, 569–581. [Google Scholar] [CrossRef]
- Byers, H.M.; Adam, M.P.; LaCroix, A.; Leary, S.E.; Cole, B.; Dobyns, W.B.; Mefford, H.C. Description of a new oncogenic mechanism for atypical teratoid rhabdoid tumors in patients with ring chromosome 22. Am. J. Med. Genet. A 2017, 173, 245–249. [Google Scholar] [CrossRef]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The central role of DNA damage in the ageing process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Endt, H.; Sprung, C.N.; Keller, U.; Gaipl, U.; Fietkau, R.; Distel, L.V. Detailed analysis of DNA repair and senescence marker kinetics over the life span of a human fibroblast cell line. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Drooger, J.C.; Hooning, M.J.; Seynaeve, C.M.; Baaijens, M.H.; Obdeijn, I.M.; Sleijfer, S.; Jager, A. Diagnostic and therapeutic ionizing radiation and the risk of a first and second primary breast cancer, with special attention for BRCA1 and BRCA2 mutation carriers: A critical review of the literature. Cancer Treat. Rev. 2015, 41, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Keller, U.; Grabenbauer, G.; Kuechler, A.; Sauer, R.; Distel, L. Radiation sensitivity testing by FISH: How many metaphases have to be analysed? Int. J. Radiat. Biol. 2004, 80, 615–620. [Google Scholar] [CrossRef]
- Keller, U.; Kuechler, A.; Liehr, T.; Muller, E.; Grabenbauer, G.; Sauer, R.; Distel, L. Impact of Various Parameters in Detecting Chromosomal Aberrations by FISH to Describe Radiosensitivity. Strahlenther. Onkol. 2004, 180, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Kuechler, A.; Neubauer, S.; Grabenbauer, G.G.; Claussen, U.; Liehr, T.; Sauer, R.; Wendt, T.G. Is 24-color FISH detection of in-vitro radiation-induced chromosomal aberrations suited to determine individual intrinsic radiosensitivity? Strahlenther. Onkol. 2002, 178, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Ziats, C.A.; Jain, L.; McLarney, B.; Vandenboom, E.; DuPont, B.R.; Rogers, C.; Sarasua, S.; Nevado, J.; Cordisco, E.L.; Phelan, K.; et al. Neurofibromatosis type 2 in Phelan-McDermid syndrome: Institutional experience and review of the literature. Eur. J. Med. Genet. 2020, 63, 104042. [Google Scholar] [CrossRef]
- Sevenet, N.; Sheridan, E.; Amram, D.; Schneider, P.; Handgretinger, R.; Delattre, O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 1999, 65, 1342–1348. [Google Scholar] [CrossRef] [Green Version]
- Boccuto, L.; Mitz, A.; Abenavoli, L.; Sarasua, S.M.; Bennett, W.; Rogers, C.; DuPont, B.; Phelan, K. Phenotypic Variability in Phelan-McDermid Syndrome and Its Putative Link to Environmental Factors. Genes 2022, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef]
- Kankuri-Tammilehto, M.; Sauna-Aho, O.; Arvio, M. Neurocognitive follow-up in adult siblings with Phelan-McDermid syndrome due to a novel SHANK3 splicing site mutation. Mol. Genet. Genom. Med. 2021, 9, e1780. [Google Scholar] [CrossRef] [PubMed]
- Vucurovic, K.; Landais, E.; Delahaigue, C.; Eutrope, J.; Schneider, A.; Leroy, C.; Kabbaj, H.; Motte, J.; Gaillard, D.; Rolland, A.C.; et al. Bipolar affective disorder and early dementia onset in a male patient with SHANK3 deletion. Eur. J. Med. Genet. 2012, 55, 625–629. [Google Scholar] [CrossRef]
- Heinen, C.D.; Schmutte, C.; Fishel, R. DNA repair and tumorigenesis: Lessons from hereditary cancer syndromes. Cancer Biol. Ther. 2002, 1, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaglia, M.C.; Giorda, R.; Beri, S.; De Agostini, C.; Novara, F.; Fichera, M.; Grillo, L.; Galesi, O.; Vetro, A.; Ciccone, R.; et al. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet. 2011, 7, e1002173. [Google Scholar] [CrossRef]
- Jeffries, A.R.; Curran, S.; Elmslie, F.; Sharma, A.; Wenger, S.; Hummel, M.; Powell, J. Molecular and phenotypic characterization of ring chromosome 22. Am. J. Med. Genet. A 2005, 137, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Luciani, J.J.; de Mas, P.; Depetris, D.; Mignon-Ravix, C.; Bottani, A.; Prieur, M.; Jonveaux, P.; Philippe, A.; Bourrouillou, G.; de Martinville, B.; et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: Cytogenetic, molecular, and clinical analyses of 32 new observations. J. Med. Genet. 2003, 40, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.F.; Rogers, R.C.; Phelan, K.; DuPont, B.R.; Collins, J.S. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, T.; Foss-Feig, J.H.; Betancur, C.; Siper, P.M.; Trelles-Thorne, M.D.P.; Halpern, D.; Frank, Y.; Lozano, R.; Layton, C.; Britvan, B.; et al. Strong evidence for genotype-phenotype correlations in Phelan-McDermid syndrome: Results from the developmental synaptopathies consortium. Hum. Mol. Genet. 2022, 31, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Nevado, J.; Garcia-Minaur, S.; Palomares-Bralo, M.; Vallespin, E.; Guillen-Navarro, E.; Rosell, J.; Bel-Fenellos, C.; Mori, M.A.; Mila, M.; Del Campo, M.; et al. Variability in Phelan-McDermid Syndrome in a Cohort of 210 Individuals. Front. Genet. 2022, 13, 652454. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Phelan McDermid Syndrome n = 20 (%) | Score (pnts) |
|---|---|---|
| sex (male; female) | 11 (55%); 9 (45%) | - |
| age (range) | 9.8 (3.5–31.8) | - |
| impaired communication receptive (minimal; light; medium; heavy) | 7 (35%); 5 (25%); 5 (25%); 3 (15%) | 3 |
| impaired communication expressive (minimal; light; medium; heavy) | 1 (5%); 9 (45%); 7 (35%); 3 (15%) | 3 |
| motor skills (normal; mild hypotonia; moderate–severe hypotonia; fine motor disturbance) | 1 (5%); 13 (65%); 2 (10%); 4 (20%) | 3 |
| cognition (not tested; below average intelligence level) | 14 (70%); 6 (30%) | 1 |
| autism (no; yes; no data) | 11 (55%); 8 (40%); 1 (5%) | 1 |
| epilepsy (no; yes; no data) | 14 (70%); 5 (25%); 1 (5%) | 1 |
| complaints of the gastrointestinal tract (no; yes; no data) | 17 (85%); 2 (10%); 1 (5%) | 1 |
| Cohort (n) | Variable | Total Cohort | Subgroup “Young“ Patients |
|---|---|---|---|
| Healthy individuals n = 218 | sex (male/female) | 93 (42.7%)/125 (57.3%) | 21 (41.2%)/30 (58.8%) |
| age (range) | 50.4 (9–81) | 25.6 (9–30) | |
| Rectal cancer n = 226 | sex (male/female) | 162 (71.7%)/64 (28.3%) | 9 (50%)/9 (50%) |
| age (range) | 63.2 (23–87) | 35.5 (23–45) | |
| Primary tumor (T1/T2/T3/T4) | 7 (3%)/28 (12.2%)/146 (64.8%)/45 (20.1%) | 1 (3%)/3 (15.2%)/12 (66.7%)/3 (15.2%) | |
| Regional lymph nodes (N0/N1) | 86 (38.2%)/140 (0%) | 6 (33.3%)/12 (66.7%) | |
| Distant metastasis (M0/M1) | 185 (81.8%)/41 (18.2%) | 15 (81.8%)/3 (18.2%) | |
| Breast cancer n = 147 | sex (female) | 147 (100%) | 27 (100%) |
| age (range) | 57.3 (28–91) | 39.1 (28–45) |
| Mean Radiation Sensitivity (B/M) | Increase Compared to PMS (%) | |||
|---|---|---|---|---|
| All | Young | All | Young | |
| PMS | 0.653 | 0.653 | - | - |
| Healthy | 0.417 | 0.377 | 56.6 | 73.2 |
| Rectal cancer | 0.434 | 0.476 | 50.5 | 37.2 |
| Breast cancer | 0.489 | 0.562 | 33.5 | 16.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jesse, S.; Kuhlmann, L.; Hildebrand, L.S.; Magelssen, H.; Schmaus, M.; Timmermann, B.; Andres, S.; Fietkau, R.; Distel, L.V. Increased Radiation Sensitivity in Patients with Phelan-McDermid Syndrome. Cells 2023, 12, 820. https://doi.org/10.3390/cells12050820
Jesse S, Kuhlmann L, Hildebrand LS, Magelssen H, Schmaus M, Timmermann B, Andres S, Fietkau R, Distel LV. Increased Radiation Sensitivity in Patients with Phelan-McDermid Syndrome. Cells. 2023; 12(5):820. https://doi.org/10.3390/cells12050820
Chicago/Turabian StyleJesse, Sarah, Lukas Kuhlmann, Laura S. Hildebrand, Henriette Magelssen, Martina Schmaus, Beate Timmermann, Stephanie Andres, Rainer Fietkau, and Luitpold V. Distel. 2023. "Increased Radiation Sensitivity in Patients with Phelan-McDermid Syndrome" Cells 12, no. 5: 820. https://doi.org/10.3390/cells12050820