


Phosphorylation of LKB1 by PDK1 Inhibits Cell Proliferation and Organ Growth by Decreased Activation of AMPK
 , , and
, , and
Abstract
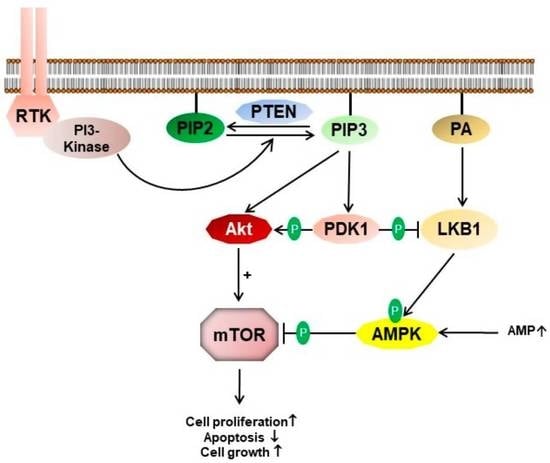
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drosophila Stocks and Genetics
2.2. Immunohistochemistry
2.3. Coimmunoprecipitation and Western Blot
2.4. In Vitro Kinase Assay
2.5. Molecular Dynamics Simulations
2.6. Statistics
3. Results
3.1. LKB1 Contains a Canonical PDK1-Binding and -Consensus Motif
3.2. Phosphorylation of LKB1 by PDK1 Does Not Affect Protein Localization In Vivo
3.3. T353 Phosphorylation Is Not Essential for Survival of the Fly, but Regulates Organism Size
3.4. Modeling of T353 Phosphorylation Reveals a Narrowed ATP-Binding Pocket
3.5. LKB1 T353 Phosphorylation Regulates Kinase Activity and Cell Growth In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kemphues, K.J.; Priess, J.R.; Morton, D.G.; Cheng, N.S. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell 1988, 52, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.L.; Morton, D.G.; Bestman, J.; Kemphues, K.J. The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development 2000, 127, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Khan, A.; St Johnston, D.; Tomlinson, I.; Martin, S.; Brenman, J.; McNeill, H. LKB1 regulates polarity remodeling and adherens junction formation in the Drosophila eye. Proc. Natl. Acad. Sci. USA 2009, 106, 8941–8946. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.G.; St Johnston, D. A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature 2003, 421, 379–384. [Google Scholar] [CrossRef]
- Bonaccorsi, S.; Mottier, V.; Giansanti, M.G.; Bolkan, B.J.; Williams, B.; Goldberg, M.L.; Gatti, M. The Drosophila Lkb1 kinase is required for spindle formation and asymmetric neuroblast division. Development 2007, 134, 2183–2193. [Google Scholar] [CrossRef] [Green Version]
- Gailite, I.; Aerne, B.L.; Tapon, N. Differential control of Yorkie activity by LKB1/AMPK and the Hippo/Warts cascade in the central nervous system. Proc. Natl. Acad. Sci. USA 2015, 112, E5169–E5178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karuman, P.; Gozani, O.; Odze, R.D.; Zhou, X.C.; Zhu, H.; Shaw, R.; Brien, T.P.; Bozzuto, C.D.; Ooi, D.; Cantley, L.C.; et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol. Cell 2001, 7, 1307–1319. [Google Scholar] [CrossRef]
- Mehenni, H.; Lin-Marq, N.; Buchet-Poyau, K.; Reymond, A.; Collart, M.A.; Picard, D.; Antonarakis, S.E. LKB1 interacts with and phosphorylates PTEN: A functional link between two proteins involved in cancer predisposing syndromes. Hum. Mol. Genet. 2005, 14, 2209–2219. [Google Scholar] [CrossRef]
- Vaahtomeri, K.; Makela, T.P. Molecular mechanisms of tumor suppression by LKB1. FEBS Lett. 2011, 585, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Muller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene 2007, 26, 7825–7832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhang, X.D.; Proud, C. Dissecting the signaling pathways that mediate cancer in PTEN and LKB1 double-knockout mice. Sci. Signal. 2015, 8, pe1. [Google Scholar] [CrossRef]
- Boudeau, J.; Baas, A.F.; Deak, M.; Morrice, N.A.; Kieloch, A.; Schutkowski, M.; Prescott, A.R.; Clevers, H.C.; Alessi, D.R. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003, 22, 5102–5114. [Google Scholar] [CrossRef] [Green Version]
- Boudeau, J.; Scott, J.W.; Resta, N.; Deak, M.; Kieloch, A.; Komander, D.; Hardie, D.G.; Prescott, A.R.; van Aalten, D.M.; Alessi, D.R. Analysis of the LKB1-STRAD-MO25 complex. J. Cell Sci. 2004, 117, 6365–6375. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, J.; Macara, I.G. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol. Biol. Cell 2008, 19, 1614–1626. [Google Scholar] [CrossRef]
- Kullmann, L.; Krahn, M.P. Controlling the master-upstream regulation of the tumor suppressor LKB1. Oncogene 2018, 37, 3045–3057. [Google Scholar] [CrossRef] [PubMed]
- Dogliotti, G.; Kullmann, L.; Dhumale, P.; Thiele, C.; Panichkina, O.; Mendl, G.; Houben, R.; Haferkamp, S.; Puschel, A.W.; Krahn, M.P. Membrane-binding and activation of LKB1 by phosphatidic acid is essential for development and tumour suppression. Nat. Commun. 2017, 8, 15747. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Park, I.H.; Wu, A.L.; Du, G.; Huang, P.; Frohman, M.A.; Walker, S.J.; Brown, H.A.; Chen, J. PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr. Biol. 2003, 13, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Strum, J.C.; Sciorra, V.A.; Daniel, L.; Bell, R.M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 1996, 271, 8472–8480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limatola, C.; Schaap, D.; Moolenaar, W.H.; van Blitterswijk, W.J. Phosphatidic acid activation of protein kinase C-zeta overexpressed in COS cells: Comparison with other protein kinase C isotypes and other acidic lipids. Biochem. J. 1994, 304 Pt 3, 1001–1008. [Google Scholar] [CrossRef]
- Toschi, A.; Lee, E.; Xu, L.; Garcia, A.; Gadir, N.; Foster, D.A. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: Competition with rapamycin. Mol. Cell Biol. 2009, 29, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Virbasius, J.V.; Song, X.; Pomerleau, D.P.; Zhan, Y.; Zhou, G.W.; Czech, M.P. Activation of the Akt-related cytokine-independent survival kinase requires interaction of its phox domain with endosomal phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA 2001, 98, 12908–12913. [Google Scholar] [CrossRef] [Green Version]
- Zelasko, J.; Czogalla, A. Selectivity of mTOR-Phosphatidic Acid Interactions Is Driven by Acyl Chain Structure and Cholesterol. Cells 2021, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Fleming, K.D.; Burke, J.E.; Leonard, T.A. Activation of the essential kinase PDK1 by phosphoinositide-driven trans-autophosphorylation. Nat. Commun. 2022, 13, 1874. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Komander, D.; Van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Truebestein, L.; Hornegger, H.; Anrather, D.; Hartl, M.; Fleming, K.D.; Stariha, J.T.B.; Pardon, E.; Steyaert, J.; Burke, J.E.; Leonard, T.A. Structure of autoinhibited Akt1 reveals mechanism of PIP3-mediated activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2101496118. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullmann, L.; Krahn, M.P. Redundant regulation of localization and protein stability of DmPar3. Cell. Mol. Life Sci. CMLS 2018, 75, 3269–3282. [Google Scholar] [CrossRef]
- Schneider, I. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 1972, 27, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Zeqiraj, E.; Filippi, B.M.; Deak, M.; Alessi, D.R.; van Aalten, D.M. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 2009, 326, 1707–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.G.; Sali, A. Modeling of loops in protein structures. Protein. Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals-a New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nose, S. A Unified Formulation of the Constant Temperature Molecular-Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Harris, T.K. Steady-state kinetic mechanism of PDK1. J. Biol. Chem. 2006, 281, 21670–21681. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Koh, H.; Kim, M.; Park, J.; Lee, S.Y.; Lee, S.; Chung, J. JNK pathway mediates apoptotic cell death induced by tumor suppressor LKB1 in Drosophila. Cell Death Differ. 2006, 13, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, F.; Lobert, V.H.; Sneeggen, M.; Jain, A.; Katheder, N.S.; Wenzel, E.M.; Schultz, S.W.; Tan, K.W.; Brech, A.; Stenmark, H.; et al. Class III phosphatidylinositol-3-OH kinase controls epithelial integrity through endosomal LKB1 regulation. Nat. Cell Biol. 2017, 19, 1412–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.L.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 1999, 22, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Pinner, S.; Sahai, E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat. Cell Biol. 2008, 10, 127–137. [Google Scholar] [CrossRef]
- Gagliardi, P.A.; Di Blasio, L.; Puliafito, A.; Seano, G.; Sessa, R.; Chianale, F.; Leung, T.; Bussolino, F.; Primo, L. PDK1-mediated activation of MRCKalpha regulates directional cell migration and lamellipodia retraction. J. Cell Biol. 2014, 206, 415–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Chikh, A.; Wheeler, A.P.; Maffucci, T.; Falasca, M. A novel regulatory mechanism links PLCgamma1 to PDK1. J. Cell Sci. 2012, 125, 3153–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Xie, Z.; Wu, Y.; Xu, J.; Dong, Y.; Zou, M.H. Protein kinase Czeta-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J. Biol. Chem. 2008, 283, 12446–12455. [Google Scholar] [CrossRef] [Green Version]
- Yamada, E.; Pessin, J.E.; Kurland, I.J.; Schwartz, G.J.; Bastie, C.C. Fyn-dependent regulation of energy expenditure and body weight is mediated by tyrosine phosphorylation of LKB1. Cell Metab. 2010, 11, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Chi, J.; Zhi, J.; Zhang, H.; Yue, D.; Zhao, J.; Li, D.; Li, Y.; Gao, M.; Guo, J. Aurora-A-mediated phosphorylation of LKB1 compromises LKB1/AMPK signaling axis to facilitate NSCLC growth and migration. Oncogene 2018, 37, 502–511. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef] [Green Version]
- Pullen, N.; Dennis, P.B.; Andjelkovic, M.; Dufner, A.; Kozma, S.C.; Hemmings, B.A.; Thomas, G. Phosphorylation and activation of p70s6k by PDK1. Science 1998, 279, 707–710. [Google Scholar] [CrossRef]
- Kobayashi, T.; Cohen, P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 1999, 339 Pt 2, 319–328. [Google Scholar] [CrossRef]
- Jensen, C.J.; Buch, M.B.; Krag, T.O.; Hemmings, B.A.; Gammeltoft, S.; Frodin, M. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 1999, 274, 27168–27176. [Google Scholar] [CrossRef] [Green Version]
- Le Good, J.A.; Ziegler, W.H.; Parekh, D.B.; Alessi, D.R.; Cohen, P.; Parker, P.J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998, 281, 2042–2045. [Google Scholar] [CrossRef] [PubMed]
- Dutil, E.M.; Toker, A.; Newton, A.C. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr. Biol. 1998, 8, 1366–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.C.; Gardiner, E.M.; Zenke, F.T.; Bohl, B.P.; Newton, A.C.; Hemmings, B.A.; Bokoch, G.M. p21-activated kinase (PAK1) is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1 (PDK1). J. Biol. Chem. 2000, 275, 41201–41209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.; Wee, Z.N.; et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirk, R.I.; Sanderson, M.R.; Lerea, K.M. Threonine phosphorylation of the beta 3 integrin cytoplasmic tail, at a site recognized by PDK1 and Akt/PKB in vitro, regulates Shc binding. J. Biol. Chem. 2000, 275, 30901–30906. [Google Scholar] [CrossRef] [Green Version]
- di Blasio, L.; Gagliardi, P.A.; Puliafito, A.; Sessa, R.; Seano, G.; Bussolino, F.; Primo, L. PDK1 regulates focal adhesion disassembly by modulating endocytosis of alphavbeta3 integrin. J. Cell Sci. 2015, 128, 863–877. [Google Scholar] [CrossRef] [Green Version]
- Deak, M.; Casamayor, A.; Currie, R.A.; Downes, C.P.; Alessi, D.R. Characterisation of a plant 3-phosphoinositide-dependent protein kinase-1 homologue which contains a pleckstrin homology domain. FEBS Lett. 1999, 451, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Milburn, C.C.; Deak, M.; Kelly, S.M.; Price, N.C.; Alessi, D.R.; Van Aalten, D.M. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem. J. 2003, 375, 531–538. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borkowsky, S.; Gass, M.; Alavizargar, A.; Hanewinkel, J.; Hallstein, I.; Nedvetsky, P.; Heuer, A.; Krahn, M.P. Phosphorylation of LKB1 by PDK1 Inhibits Cell Proliferation and Organ Growth by Decreased Activation of AMPK. Cells 2023, 12, 812. https://doi.org/10.3390/cells12050812
Borkowsky S, Gass M, Alavizargar A, Hanewinkel J, Hallstein I, Nedvetsky P, Heuer A, Krahn MP. Phosphorylation of LKB1 by PDK1 Inhibits Cell Proliferation and Organ Growth by Decreased Activation of AMPK. Cells. 2023; 12(5):812. https://doi.org/10.3390/cells12050812
Chicago/Turabian StyleBorkowsky, Sarah, Maximilian Gass, Azadeh Alavizargar, Johannes Hanewinkel, Ina Hallstein, Pavel Nedvetsky, Andreas Heuer, and Michael P. Krahn. 2023. "Phosphorylation of LKB1 by PDK1 Inhibits Cell Proliferation and Organ Growth by Decreased Activation of AMPK" Cells 12, no. 5: 812. https://doi.org/10.3390/cells12050812