
Enzymology of Ca2+-Mobilizing Second Messengers Derived from NAD: From NAD Glycohydrolases to (Dual) NADPH Oxidases

{kind=link}
{kind=link}
Abstract
:1. Main Text
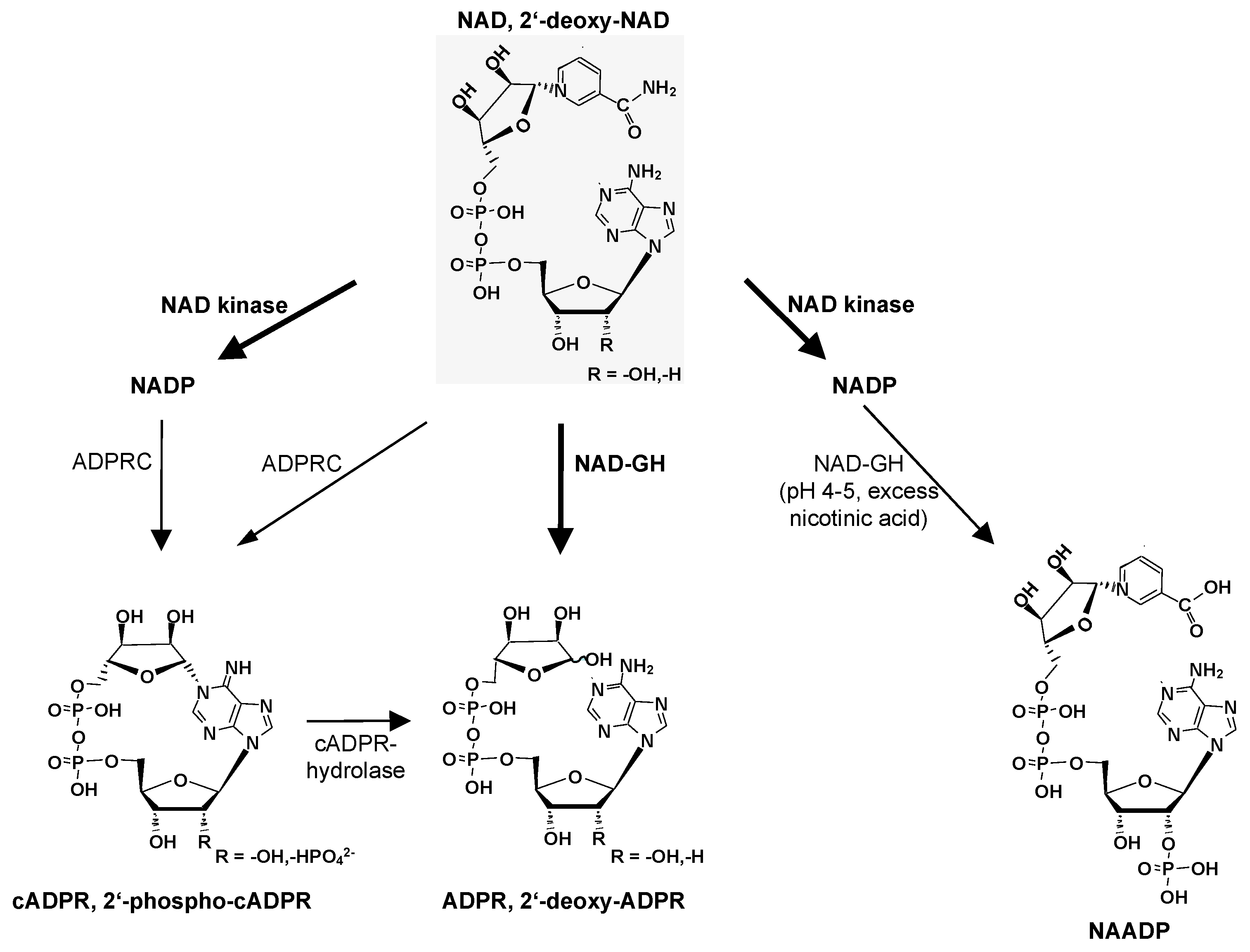
2. NAD-GHs
3. CD38
4. SARM1
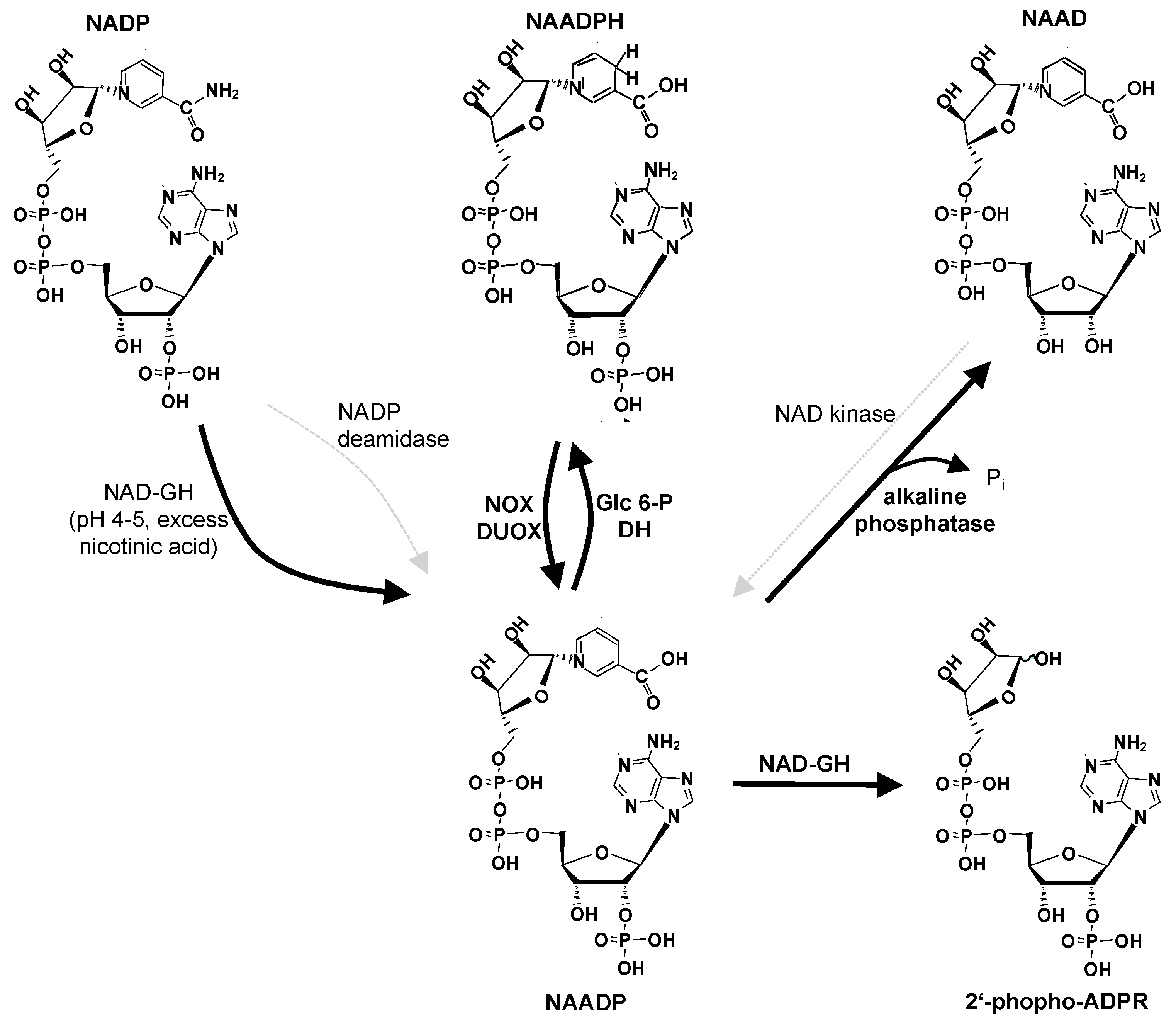
5. NAADPH/NAADP Redox Cycle: (Dual) NADPH Oxidases (NOX/DUOX) and Glucose 6-Phosphate Dehydrogenase (G6P-DH)
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and Degradation of Cyclic ADP-Ribose by NAD-GHs. Science 1993, 261, 1330–1333. [Google Scholar] [CrossRef]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [CrossRef]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [CrossRef]
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef]
- Lee, H.C.; Aarhus, R. A Derivative of NADP Mobilizes Calcium Stores Insensitive to Inositol Trisphosphate and Cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.E.; Walseth, T.F.; Lee, H.C. Formation and Hydrolysis of Cyclic ADP-Ribose Catalyzed by Lymphocyte Antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD + Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef] [Green Version]
- Terhorst, C.; Van Agthoven, A.; LeClair, K.; Snow, P.; Reinherz, E.; Schlossman, S. Biochemical studies of the human thymocyte cell-surface antigens T6, T9 and T10. Cell 1981, 23, 771–780. [Google Scholar] [CrossRef]
- Zocchi, E.; Franco, L.; Guida, L.; Benatti, U.; Bargellesi, A.; Malavasi, F.; Lee, H.; Deflora, A. A Single Protein Immunologically Identified as CD38 Displays NAD+ Glycohydrolase, ADP-Ribosyl Cyclase and Cyclic ADP-Ribose Hydrolase Activities at the Outer Surface of Human Erythrocytes. Biochem. Biophys. Res. Commun. 1993, 196, 1459–1465. [Google Scholar] [CrossRef]
- Lee, H.; Zocchi, E.; Guida, L.; Franco, L.; Benatti, U.; Deflora, A. Production and Hydrolysis of Cyclic ADP-Ribose at the Outer Surface of Human Erythrocytes. Biochem. Biophys. Res. Commun. 1993, 191, 639–645. [Google Scholar] [CrossRef]
- De Flora, A.; Guida, L.; Franco, L.; Zocchi, E. The CD38/cyclic ADP-ribose system: A topological paradox. Int. J. Biochem. Cell Biol. 1997, 29, 1149–1166. [Google Scholar] [CrossRef]
- Flora, A.; Franco, L.; Guida, L.; Bruzzone, S.; Zocchi, E. Ectocellular CD38-catalyzed synthesis and intracellular Ca2+-mobilizing activity of cyclic ADP-ribose. Cell Biochem. Biophys. 1998, 28, 45–62. [Google Scholar] [CrossRef]
- Franco, L.; Guida, L.; Bruzzone, S.; Zocchi, E.; Usai, C.; De Flora, A. The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP--ribose across membranes. FASEB J. 1998, 12, 1507–1520. [Google Scholar] [CrossRef]
- Zocchi, E.; Usai, C.; Guida, L.; Franco, L.; Bruzzone, S.; Passalacqua, M.; DE Flora, A. Ligand--induced internalization of CD38 results in intracellular Ca2+ mobilization: Role of NAD+ transport across cell membranes. FASEB J. 1999, 13, 273–283. [Google Scholar] [CrossRef]
- Bruzzone, S.; Franco, L.; Guida, L.; Zocchi, E.; Contini, P.; Bisso, A.; Usai, C.; De Flora, A. A Self-restricted CD38-connexin 43 Cross-talk Affects NAD+ and Cyclic ADP-ribose Metabolism and Regulates Intracellular Calcium in 3T3 Fibroblasts. J. Biol. Chem. 2001, 276, 48300–48308. [Google Scholar] [CrossRef] [Green Version]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef]
- Franco, L.; Bruzzone, S.; Song, P.; Guida, L.; Zocchi, E.; Walseth, T.F.; Crimi, E.; Usai, C.; De Flora, A.; Brusasco, V. Extracellular cyclic ADP-ribose potentiates ACh-induced contraction in bovine tracheal smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L98–L106. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.; Zocchi, E.; Usai, C.; Guida, L.; Bruzzone, S.; Costa, A.; De Flora, A. Paracrine roles of NAD+and cyclic ADP-ribose in increasing intracellular calcium and enhancing cell proliferation of 3T3 fibroblasts. J. Biol. Chem. 2001, 276, 21642–21648. [Google Scholar] [CrossRef] [Green Version]
- Zocchi, E.; Podestà, M.; Pitto, A.; Usai, C.; Bruzzone, S.; Franco, L.; Guida, L.; Bacigalupo, A.; De Flora, A. Paracrinally stimulated expansion of early human hemopoietic progenitors by stroma-generated cyclic ADP-ribose. FASEB J. 2001, 15, 1610–1612. [Google Scholar] [CrossRef]
- Yilmaz, H.; Katajisto, P.; Lamming, D.W.; Gültekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Guida, L.; Bruzzone, S.; Sturla, L.; Franco, L.; Zocchi, E.; De Flora, A. Equilibrative and Concentrative Nucleoside Transporters Mediate Influx of Extracellular Cyclic ADP-Ribose into 3T3 Murine Fibroblasts. J. Biol. Chem. 2002, 277, 47097–47105. [Google Scholar] [CrossRef] [Green Version]
- Guida, L.; Franco, L.; Bruzzone, S.; Sturla, L.; Zocchi, E.; Basile, G.; Usai, C.; De Flora, A. Concentrative Influx of Functionally Active Cyclic ADP-ribose in Dimethyl Sulfoxide-differentiated HL-60 Cells. J. Biol. Chem. 2004, 279, 22066–22075. [Google Scholar] [CrossRef] [Green Version]
- Podestà, M.; Benvenuto, F.; Pitto, A.; Figari, O.; Bacigalupo, A.; Bruzzone, S.; Guida, L.; Franco, L.; Paleari, L.; Bodrato, N.; et al. Concentrative Uptake of Cyclic ADP-ribose Generated by BST-1+ Stroma Stimulates Proliferation of Human Hematopoietic Progenitors. J. Biol. Chem. 2005, 280, 5343–5349. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Anglada, M.; Cano-Soldado, P.; Errasti-Murugarren, E.; Casado, F.J. SLC28 genes and concentrative nucleoside transporter (CNT) proteins. Xenobiotica 2008, 38, 972–994. [Google Scholar] [CrossRef]
- Astigiano, C.; Benzi, A.; Laugieri, M.E.; Piacente, F.; Sturla, L.; Guida, L.; Bruzzone, S.; De Flora, A. Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells 2022, 11, 2637. [Google Scholar] [CrossRef]
- Zhong, X.; Ma, H. Targeting CD38 for acute leukemia. Front. Oncol. 2022, 12, 1007783. [Google Scholar] [CrossRef]
- Hambach, J.; Mann, A.M.; Bannas, P.; Koch-Nolte, F. Targeting multiple myeloma with nanobody-based heavy chain antibodies, bispecific killer cell engagers, chimeric antigen receptors, and nanobody-displaying AAV vectors. Front. Immunol. 2022, 13, 1005800. [Google Scholar] [CrossRef]
- Gao, L.; Du, X.; Li, J.; Qin, F.X.-F. Evolving roles of CD38 metabolism in solid tumour microenvironment. Br. J. Cancer 2023, 128, 492–504. [Google Scholar] [CrossRef]
- Kato, I.; Yamamoto, Y.; Fujimura, M.; Noguchi, N.; Takasawa, S.; Okamoto, H. CD38 Disruption Impairs Glucose-induced Increases in Cyclic ADP-ribose, [Ca2+], and Insulin Secretion. J. Biol. Chem. 1999, 274, 1869–1872. [Google Scholar] [CrossRef] [Green Version]
- Fukushi, Y.; Kato, I.; Takasawa, S.; Sasaki, T.; Ong, B.H.; Sato, M.; Ohsaga, A.; Sato, K.; Shirato, K.; Okamoto, H.; et al. Identification of Cyclic ADP-ribose-dependent Mechanisms in Pancreatic Muscarinic Ca2+ Signaling Using CD38 Knockout Mice. J. Biol. Chem. 2001, 276, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Mitsui-Saito, M.; Kato, I.; Takasawa, S.; Okamoto, H.; Yanagisawa, T. CD38 Gene Disruption Inhibits the Contraction Induced by α-Adrenoceptor Stimulation in Mouse Aorta. J. Veter. Med. Sci. 2003, 65, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Rah, S.-Y.; Park, K.-H.; Han, M.-K.; Im, M.-J.; Kim, U.-H. Activation of CD38 by Interleukin-8 Signaling Regulates Intracellular Ca2+ Level and Motility of Lymphokine-activated Killer Cells. J. Biol. Chem. 2005, 280, 2888–2895. [Google Scholar] [CrossRef] [Green Version]
- Rah, S.-Y.; Mushtaq, M.; Nam, T.-S.; Kim, S.H.; Kim, U.-H. Generation of Cyclic ADP-ribose and Nicotinic Acid Adenine Dinucleotide Phosphate by CD38 for Ca2+ Signaling in Interleukin-8-treated Lymphokine-activated Killer Cells. J. Biol. Chem. 2010, 285, 21877–21887. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Cho, B.H.; Kim, U.-H. CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells: Attenuation of hepatic fibrosis by CD38 ablation. J. Biol. Chem. 2010, 285, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Cockayne, D.A.; Muchamuel, T.; Grimaldi, J.C.; Muller-Steffner, H.; Randall, T.D.; Lund, F.E.; Murray, R.; Schuber, F.; Howard, M.C. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood 1998, 92, 1324–1333. [Google Scholar] [CrossRef]
- Lund, F.E.; Muller-Steffner, H.M.; Yu, N.; Stout, C.D.; Schuber, F.; Howard, M.C. CD38 signaling in B lymphocytes is controlled by its ectodomain but occurs independently of enzymatically generated ADP-ribose or cyclic ADP-ribose. J. Immunol. 1999, 162, 2693–2702. [Google Scholar] [CrossRef]
- Partida-Sánchez, S.; Cockayne, D.A.; Monard, S.; Jacobson, E.L.; Oppenheimer, N.; Garvy, B.; Kusser, K.; Goodrich, S.; Howard, M.; Harmsen, A.; et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001, 7, 1209–1216. [Google Scholar] [CrossRef]
- Partida-Sanchez, S.; Gasser, A.; Fliegert, R.; Siebrands, C.C.; Dammermann, W.; Shi, G.; Mousseau, B.J.; Sumoza-Toledo, A.; Bhagat, H.; Walseth, T.F.; et al. Chemotaxis of Mouse Bone Marrow Neutrophils and Dendritic Cells Is Controlled by ADP-Ribose, the Major Product Generated by the CD38 Enzyme Reaction. J. Immunol. 2007, 179, 7827–7839. [Google Scholar] [CrossRef] [Green Version]
- Berthelier, V.; Tixier, J.-M.; Muller-Steffner, H.; Schuber, F.; Deterre, P. Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem. J. 1998, 330, 1383–1390. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, K.; Mayr, G.W.; Guse, A.H. Assay for ADP-Ribosyl Cyclase by Reverse-Phase High-Performance Liquid Chromatography. Anal. Biochem. 2001, 299, 218–226. [Google Scholar] [CrossRef]
- Graeff, R.; Liu, Q.; Kriksunov, I.A.; Kotaka, M.; Oppenheimer, N.; Hao, Q.; Lee, H.C. Mechanism of Cyclizing NAD to Cyclic ADP-ribose by ADP-ribosyl Cyclase and CD38. J. Biol. Chem. 2009, 284, 27629–27636. [Google Scholar] [CrossRef] [Green Version]
- Kirchberger, T.; Guse, A.H. Measuring CD38 (ADP-Ribosyl Cyclase/Cyclic ADP-Ribose Hydrolase) Activity by Reverse-Phase HPLC. Cold Spring Harb. Protoc. 2013, 2013, 569–573. [Google Scholar] [CrossRef]
- Rah, S.-Y.; Kwak, J.-Y.; Chung, Y.-J.; Kim, U.-H. ADP-ribose/TRPM2-mediated Ca2+ signaling is essential for cytolytic degranulation and antitumor activity of natural killer cells. Sci. Rep. 2015, 5, srep09482. [Google Scholar] [CrossRef] [Green Version]
- Fliegert, R.; Bauche, A.; Pérez, A.-M.W.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.J.; Lam, C.M.C.; Lee, H.C. The Membrane-Bound Enzyme CD38 Exists in Two Opposing Orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef]
- Park, D.-R.; Nam, T.-S.; Kim, Y.-W.; Bae, Y.S.; Kim, U.-H. Oxidative activation of type III CD38 by NADPH oxidase–derived hydrogen peroxide in Ca2+ signaling. FASEB J. 2018, 33, 3404–3419. [Google Scholar] [CrossRef]
- Muller-Steffner, H.M.; Malver, O.; Hosie, L.; Oppenheimer, N.J.; Schuber, F. Slow-binding inhibition of NAD+ glycohydrolase by arabino analogues of beta-NAD. J. Biol. Chem. 1992, 267, 9606–9611. [Google Scholar] [CrossRef]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Hon, C.L. ADP-ribosyl Cyclase and CD38 Catalyze the Synthesis of a Calcium-mobilizing Metabolite from NADP+. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Li, T.; Li, Y.; Xu, G.J.; Deng, Q.W.; Chen, Y.J.; Hou, Y.N.; Lee, H.C.; Zhao, Y.J. CD38 produces nicotinic acid adenosine dinucleotide phosphate in the lysosome. J. Biol. Chem. 2018, 293, 8151–8160. [Google Scholar] [CrossRef] [Green Version]
- Nam, T.; Park, D.; Rah, S.; Woo, T.; Chung, H.T.; Brenner, C.; Kim, U. Interleukin--8 drives CD38 to form NAADP from NADP+ and NAAD in the endolysosomes to mobilize Ca2+ and effect cell migration. FASEB J. 2020, 34, 12565–12576. [Google Scholar] [CrossRef]
- Zhang, F.; Xia, M.; Li, P.-L. Lysosome-dependent Ca2+ release response to Fas activation in coronary arterial myocytes through NAADP: Evidence from CD38 gene knockouts. Am. J. Physiol. Physiol. 2010, 298, C1209–C1216. [Google Scholar] [CrossRef] [Green Version]
- Cosker, F.; Cheviron, N.; Yamasaki, M.; Menteyne, A.; Lund, F.E.; Moutin, M.-J.; Galione, A.; Cancela, J.-M. The Ecto-enzyme CD38 Is a Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Synthase That Couples Receptor Activation to Ca2+ Mobilization from Lysosomes in Pancreatic Acinar Cells. J. Biol. Chem. 2010, 285, 38251–38259. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.K.; Bolton, E.L.; Cortopassi, W.A.; Wang, Y.; O’Brien, F.; Maciejewska, M.; Jacobson, M.P.; Garnham, C.; Ruas, M.; Parrington, J.; et al. Synthesis of the Ca2+-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in β-adrenoceptor signaling. J. Biol. Chem. 2017, 292, 13243–13257. [Google Scholar] [CrossRef] [Green Version]
- Soares, S.; Thompson, M.; White, T.; Isbell, A.; Yamasaki, M.; Prakash, Y.; Lund, F.E.; Galione, A.; Chini, E.N. NAADP as a second messenger: Neither CD38 nor base-exchange reaction are necessary for in vivo generation of NAADP in myometrial cells. Am. J. Physiol. Physiol. 2007, 292, C227–C239. [Google Scholar] [CrossRef] [Green Version]
- Schmid, F.; Bruhn, S.; Weber, K.; Mittrücker, H.-W.; Guse, A.H. CD38: A NAADP degrading enzyme. FEBS Lett. 2011, 585, 3544–3548. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Doan, R.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Doan, R.A.; Cheng, G.C.; Cetinkaya-Fisgin, A.; Huang, S.X.; Höke, A.; Milbrandt, J.; DiAntonio, A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. J. Clin. Investig. 2019, 4, e129920. [Google Scholar] [CrossRef] [Green Version]
- Cetinkaya-Fisgin, A.; Luan, X.; Reed, N.; Jeong, Y.E.; Oh, B.C.; Hoke, A. Cisplatin induced neurotoxicity is mediated by Sarm1 and calpain activation. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Marion, C.M.; McDaniel, D.P.; Armstrong, R.C. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp. Neurol. 2019, 321, 113040. [Google Scholar] [CrossRef]
- Ko, K.W.; Milbrandt, J.; DiAntonio, A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 2020, 219, e201912047. [Google Scholar] [CrossRef]
- DiAntonio, A.; Milbrandt, J.; Figley, M.D. The SARM1 TIR NADase: Mechanistic Similarities to Bacterial Phage Defense and Toxin-Antitoxin Systems. Front. Immunol. 2021, 12, 752898. [Google Scholar] [CrossRef]
- Zhao, Z.Y.; Xie, X.J.; Li, W.H.; Liu, J.; Chen, Z.; Zhang, B.; Li, T.; Li, S.L.; Lu, J.G.; Zhang, L.; et al. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. Iscience 2019, 15, 452–466. [Google Scholar] [CrossRef] [Green Version]
- Angeletti, C.; Amici, A.; Gilley, J.; Loreto, A.; Trapanotto, A.G.; Antoniou, C.; Merlini, E.; Coleman, M.P.; Orsomando, G. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. Iscience 2022, 25, 103812. [Google Scholar] [CrossRef]
- Shawl, A.I.; Park, K.-H.; Kim, U.-H. Insulin receptor signaling for the proliferation of pancreatic β-cells: Involvement of Ca2+ second messengers, IP3, NAADP and cADPR. Islets 2009, 1, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Guse, A.H.; Da Silva, C.P.; Berg, I.; Skapenko, A.L.; Weber, K.; Heyer, P.; Hohenegger, M.; Ashamu, G.A.; Schulze-Koops, H.; Potter, B.V.L.; et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 1999, 398, 70–73. [Google Scholar] [CrossRef]
- Hou, Y.N.; Cai, Y.; Li, W.H.; He, W.M.; Zhao, Z.Y.; Zhu, W.J.; Wang, Q.; Mai, X.; Liu, J.; Lee, H.C.; et al. A conformation-specific nanobody targeting the nicotinamide mononucleotide-activated state of SARM1. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Waller, T.J.; Collins, C.A. Multifaceted roles of SARM1 in axon degeneration and signaling. Front. Cell. Neurosci. 2022, 16, 958900. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 Is Required for Activation of an Injury-Induced Axon Death Pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Imai, T. Purification and Properties of Nicotinamide Mononucleotide Amidohydrolase from Azotobacter vinelandii. J. Biochem. 1973, 73, 139–153. [Google Scholar] [CrossRef]
- Foster, J.W.; Baskowsky-Foster, A.M. Pyridine nucleotide cycle of Salmonella typhimurium: In vivo recycling of nicotinamide adenine dinucleotide. J. Bacteriol. 1980, 142, 1032–1035. [Google Scholar] [CrossRef] [Green Version]
- Hillyard, D.; Rechsteiner, M.; Manlapaz-Ramos, P.; Imperial, J.S.; Cruz, L.J.; Olivera, B.M. The pyridine nucleotide cycle. Studies in Escherichia coli and the human cell line D98/AH2. J. Biol. Chem. 1981, 256, 8491–8497. [Google Scholar] [CrossRef]
- Foster, J.W.; Brestel, C. NAD metabolism in Vibrio cholerae. J. Bacteriol. 1982, 149, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Galeazzi, L.; Bocci, P.; Amici, A.; Brunetti, L.; Ruggieri, S.; Romine, M.; Reed, S.; Osterman, A.L.; Rodionov, D.; Sorci, L.; et al. Identification of Nicotinamide Mononucleotide Deamidase of the Bacterial Pyridine Nucleotide Cycle Reveals a Novel Broadly Conserved Amidohydrolase Family. J. Biol. Chem. 2011, 286, 40365–40375. [Google Scholar] [CrossRef] [Green Version]
- Lerner, F.; Niere, M.; Ludwig, A.; Ziegler, M. Structural and Functional Characterization of Human NAD Kinase. Biochem. Biophys. Res. Commun. 2001, 288, 69–74. [Google Scholar] [CrossRef]
- Billington, R.A.; Thuring, J.W.; Conway, S.J.; Packman, L.; Holmes, A.B.; Genazzani, A.A. Production and characterization of reduced NAADP (nicotinic acid-adenine dinucleotide phosphate). Biochem. J. 2004, 378, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Buvelot, H.; Jaquet, V.; Krause, K.-H. Mammalian NADPH Oxidases. In NADPH Oxidases. Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1982, pp. 17–36. [Google Scholar] [CrossRef]
- Gu, F.; Krüger, A.; Roggenkamp, H.G.; Alpers, R.; Lodygin, D.; Jaquet, V.; Möckl, F.; C., L.C.H.; Winterberg, K.; Bauche, A.; et al. Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca2+ signaling during T cell activation. Sci. Signal. 2021, 14, eabe3800. [Google Scholar] [CrossRef]
- Wolf, I.M.A.; Diercks, B.-P.; Gattkowski, E.; Czarniak, F.; Kempski, J.; Werner, R.; Schetelig, D.; Mittrücker, H.-W.; Schumacher, V.; von Osten, M.; et al. Frontrunners of T cell activation: Initial, localized Ca2+ signals mediated by NAADP and the type 1 ryanodine receptor. Sci. Signal. 2015, 8, ra102. [Google Scholar] [CrossRef]
- Grasberger, H.; Refetoff, S. Identification of the Maturation Factor for Dual Oxidase: Evolution of an eukaryotic operon equivalent. J. Biol. Chem. 2006, 281, 18269–18272. [Google Scholar] [CrossRef] [Green Version]
- Grasberger, H.; De Deken, X.; Mayo, O.B.; Raad, H.; Weiss, M.; Liao, X.-H.; Refetoff, S. Mice Deficient in Dual Oxidase Maturation Factors Are Severely Hypothyroid. Mol. Endocrinol. 2012, 26, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Gasser, A.; Bruhn, S.; Guse, A.H. Second Messenger Function of Nicotinic Acid Adenine Dinucleotide Phosphate Revealed by an Improved Enzymatic Cycling Assay. J. Biol. Chem. 2006, 281, 16906–16913. [Google Scholar] [CrossRef] [Green Version]
- Wehage, E.; Eisfeld, J.; Heiner, I.; Jüngling, E.; Zitt, C.; Lückhoff, A. Activation of the Cation Channel Long Transient Receptor Potential Channel 2 (LTRPC2) by Hydrogen Peroxide. J. Biol. Chem. 2002, 277, 23150–23156. [Google Scholar] [CrossRef] [Green Version]
- Roscoe, J.; Sevier, C. Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum. Cells 2020, 9, 2314. [Google Scholar] [CrossRef]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef]
- Schmid, F.; Fliegert, R.; Westphal, T.; Bauche, A.; Guse, A.H. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Degradation by Alkaline Phosphatase. J. Biol. Chem. 2012, 287, 32525–32534. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guse, A.H. Enzymology of Ca2+-Mobilizing Second Messengers Derived from NAD: From NAD Glycohydrolases to (Dual) NADPH Oxidases. Cells 2023, 12, 675. https://doi.org/10.3390/cells12040675
Guse AH. Enzymology of Ca2+-Mobilizing Second Messengers Derived from NAD: From NAD Glycohydrolases to (Dual) NADPH Oxidases. Cells. 2023; 12(4):675. https://doi.org/10.3390/cells12040675
Chicago/Turabian StyleGuse, Andreas H. 2023. "Enzymology of Ca2+-Mobilizing Second Messengers Derived from NAD: From NAD Glycohydrolases to (Dual) NADPH Oxidases" Cells 12, no. 4: 675. https://doi.org/10.3390/cells12040675