
Inhibition of Soluble Epoxide Hydrolase Does Not Promote or Aggravate Pulmonary Hypertension in Rats
 , , , , , , ,
, , , , , , ,  and
and 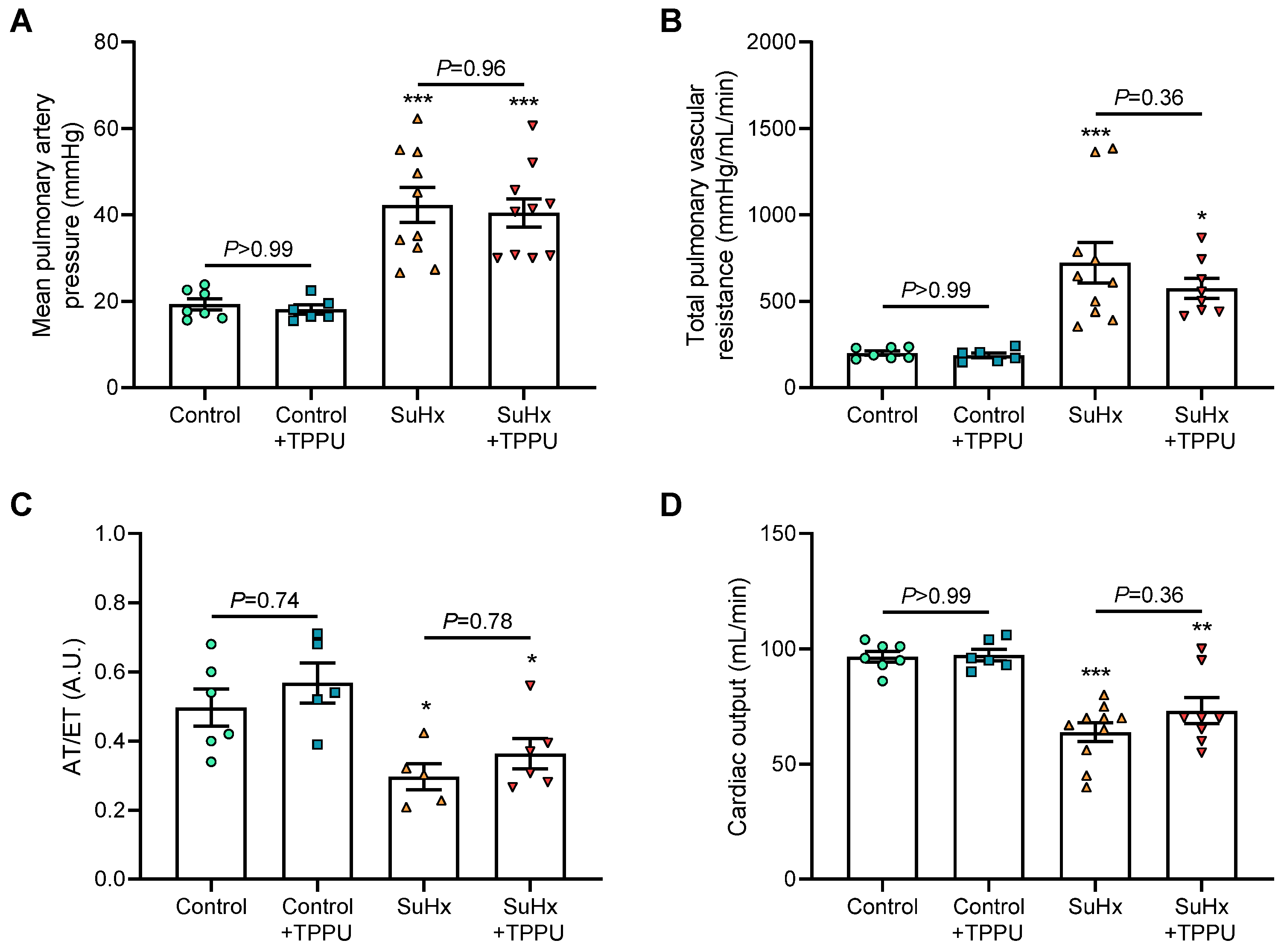{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Cardiovascular Parameters Investigated
2.3. Cardiovascular Parameters Investigated
2.3.1. Chemicals and Reagents
2.3.2. Sample Preparation
2.3.3. LC-MS/MS Conditions
2.4. Proliferation of Human PA-SMCs
2.5. Statistical Analyses
3. Results
3.1. sEH Inhibition Does Not Alter Hemodynamic and Pulmonary Hemodynamic Parameters in Healthy Sprague–Dawley Rats
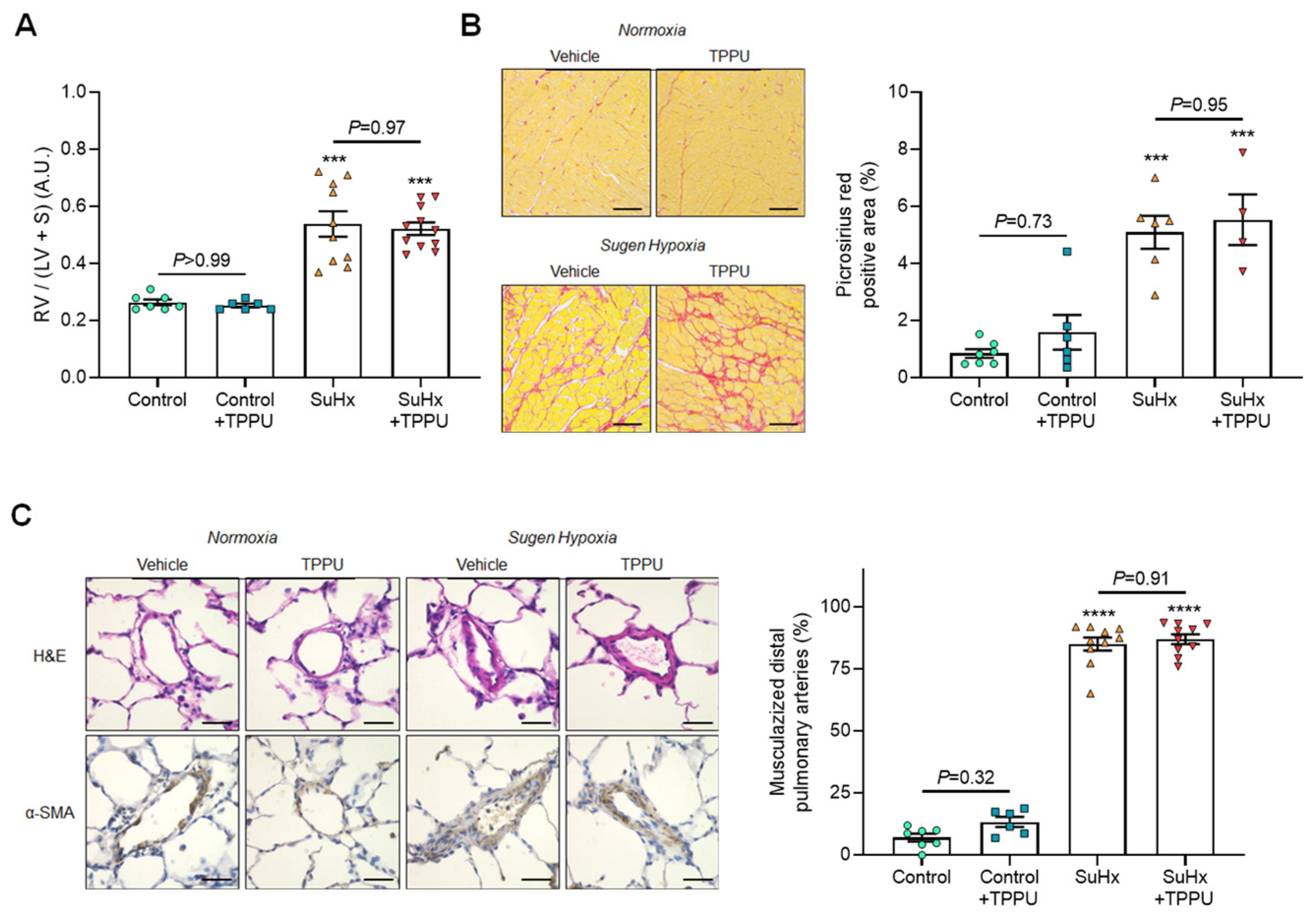
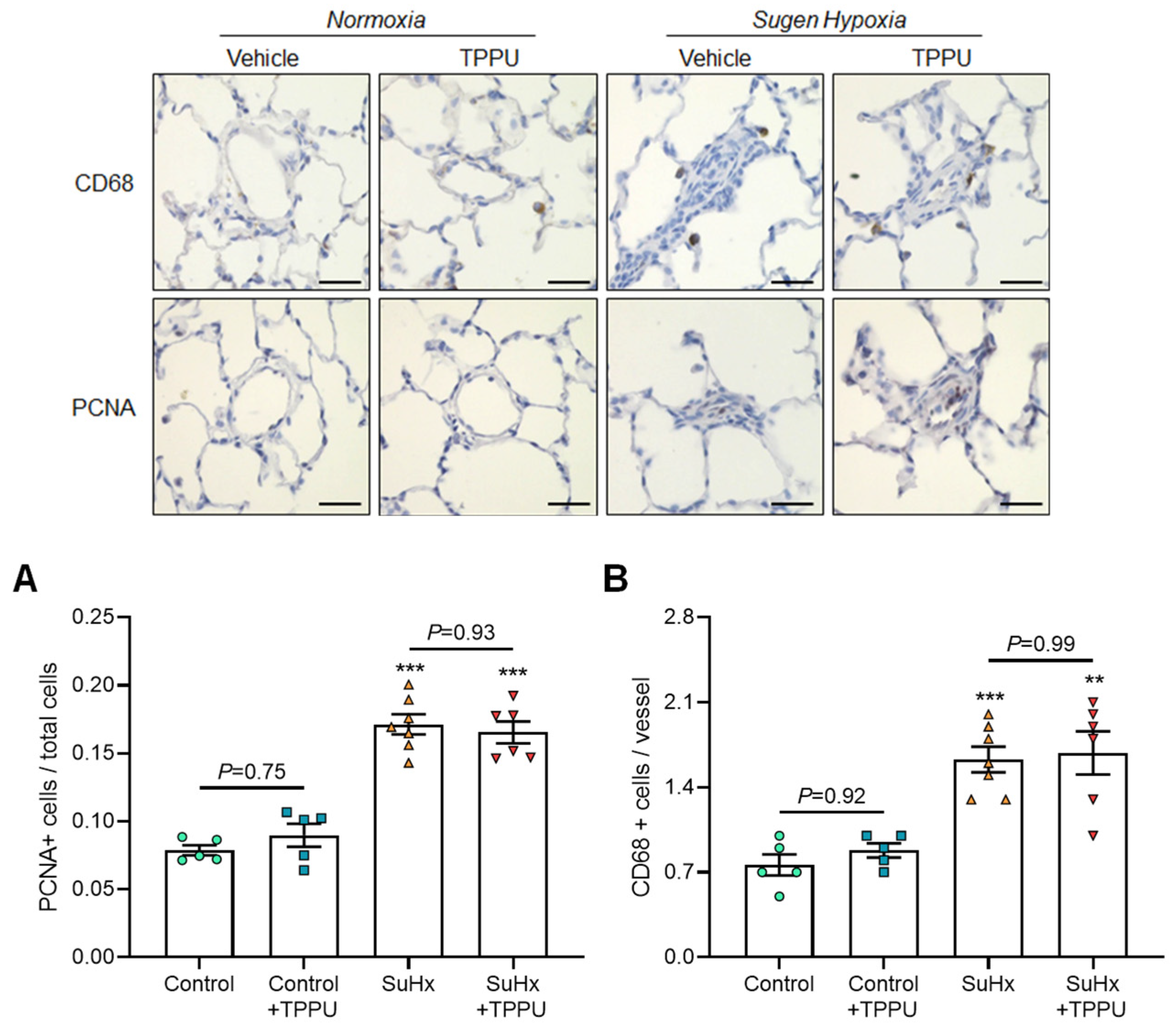
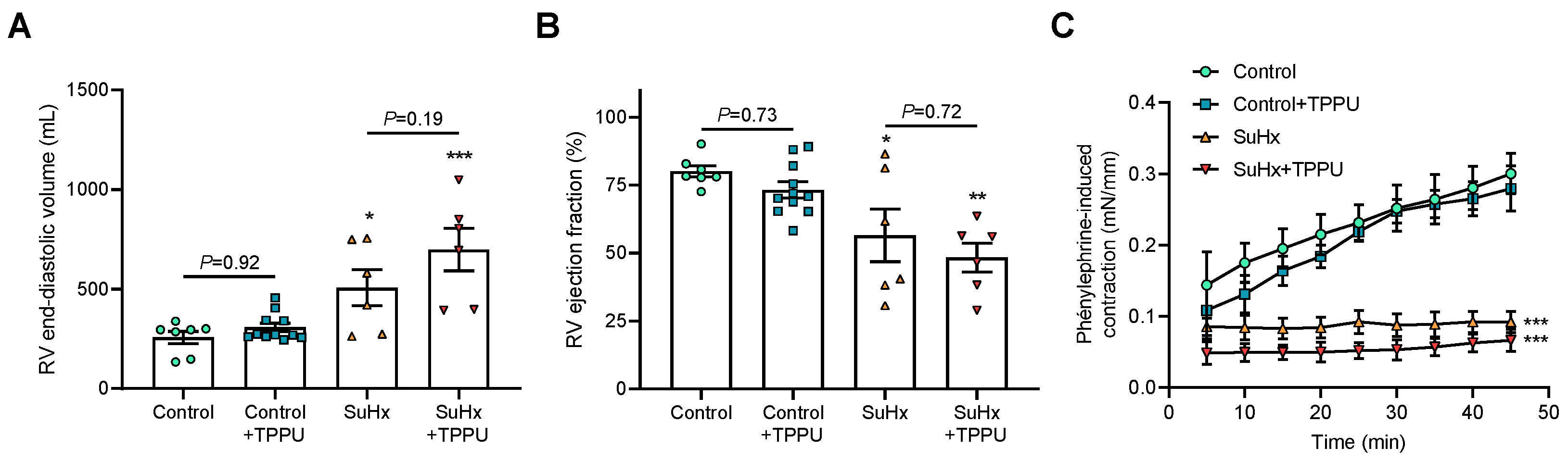
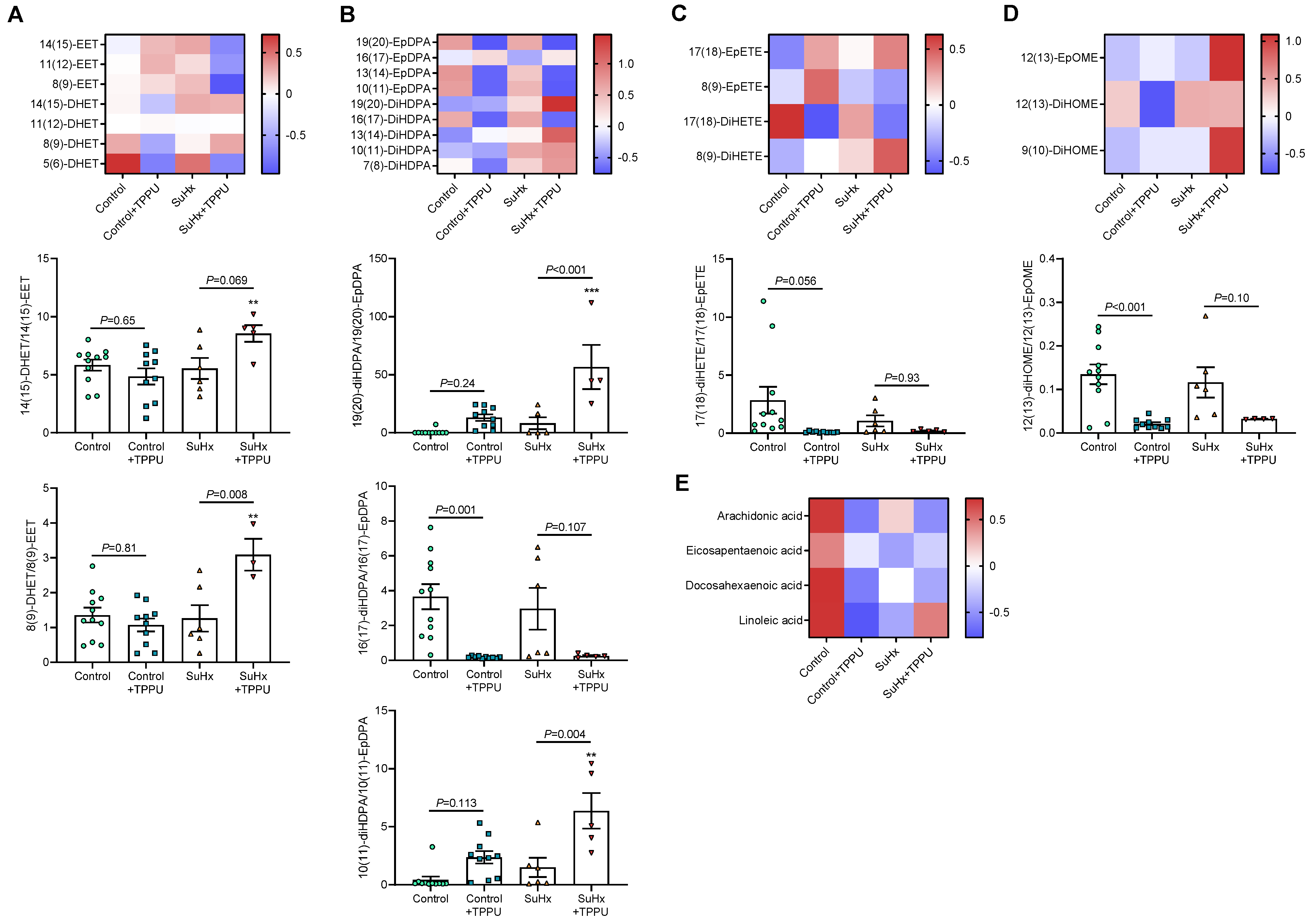
3.2. sEH Inhibition Does Not Alter the Progression of PH in the SUGEN + Hypoxia (SuHx) Rat Model
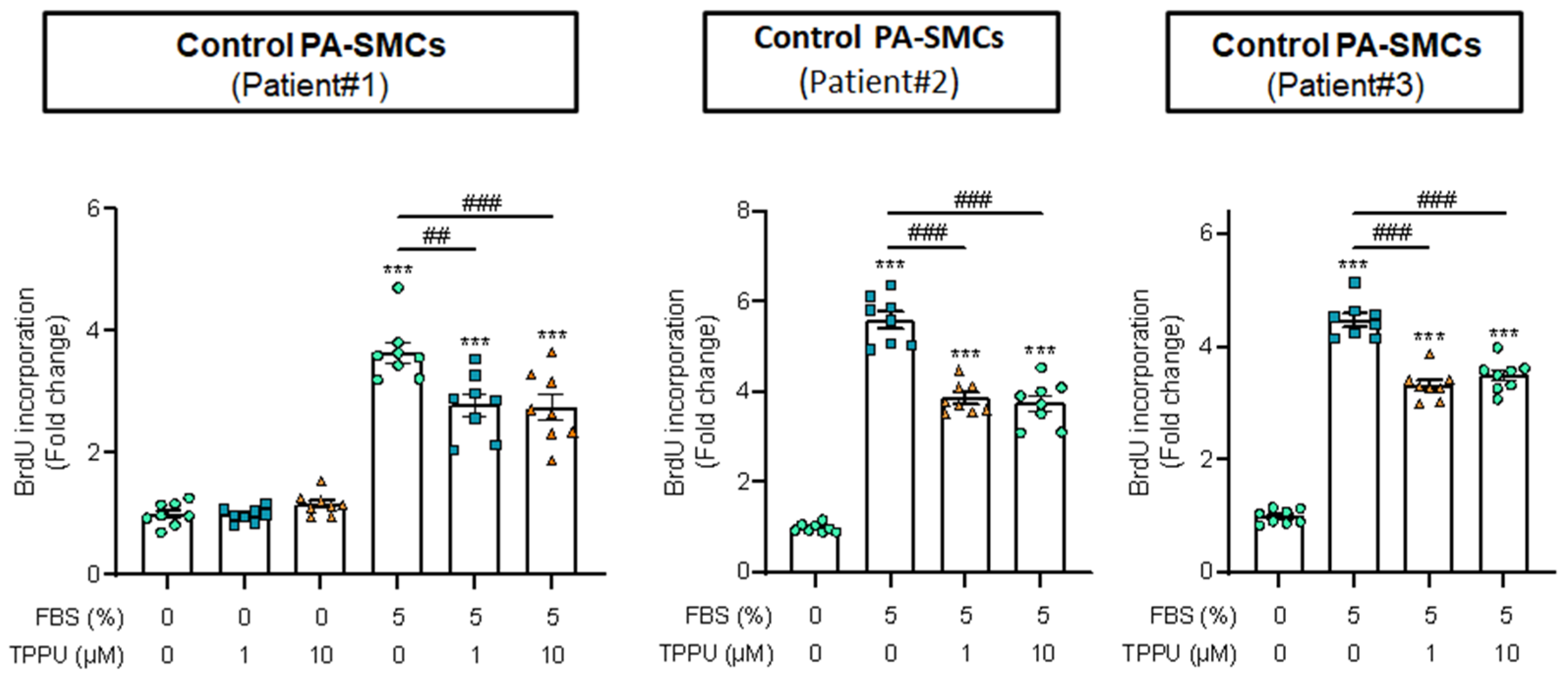
3.3. sEH Inhibition Attenuates Proliferation of Cultured Human PA-SMCs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morisseau, C.; Hammock, B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellien, J.; Joannides, R.; Richard, V.; Thuillez, C. Modulation of cytochrome-derived epoxyeicosatrienoic acids pathway: A promising pharmacological approach to prevent endothelial dysfunction in cardiovascular diseases? Pharmacol. Ther. 2011, 131, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lazaar, A.L.; Yang, L.; Boardley, R.L.; Goyal, N.S.; Robertson, J.; Baldwin, S.J.; Newby, D.E.; Wilkinson, I.B.; Tal-Singer, R.; Mayer, R.J.; et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 2016, 81, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Whitcomb, R.; MacIntyre, E.; Tran, V.; Do, Z.N.; Sabry, J.; Patel, D.V.; Anandan, S.K.; Gless, R.; Webb, H.K. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 2012, 52, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Fleming, I. Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: Central role of transient receptor potential C6 channels. J. Cardiovasc. Pharmacol. 2011, 57, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Kandhi, S.; Froogh, G.; Qin, J.; Luo, M.; Wolin, M.S.; Huang, A.; Sun, D. EETs Elicit Direct Increases in Pulmonary Arterial Pressure in Mice. Am. J. Hypertens. 2016, 29, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Pokreisz, P.; Fleming, I.; Kiss, L.; Barbosa-Sicard, E.; Fisslthaler, B.; Falck, J.; Hammock, B.D.; Kim, I.-H.; Szelid, Z.; Vermeersch, P.; et al. Cytochrome P450 epoxygenase gene function in hypoxic pulmonary vasoconstriction and pulmonary vascular remodeling. Hypertension 2006, 47, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef]
- Keserü, B.; Barbosa-Sicard, E.; Popp, R.; Fisslthaler, B.; Dietrich, A.; Gudermann, T.; Hammock, B.D.; Falck, J.R.; Weissmann, N.; Busse, R.; et al. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J. 2008, 22, 4306–4315. [Google Scholar] [CrossRef] [Green Version]
- Revermann, M.; Barbosa-Sicard, E.; Dony, E.; Schermuly, R.T.; Morisseau, C.; Geisslinger, G.; Fleming, I.; Hammock, B.D.; Brandes, R.P. Inhibition of the soluble epoxide hydrolase attenuates monocrotaline-induced pulmonary hypertension in rats. J. Hypertens. 2009, 27, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Bordenave, J.; Thuillet, R.; Tu, L.; Phan, C.; Cumont, A.; Marsol, C.; Huertas, A.; Savale, L.; Hibert, M.; Galzi, J.-L.; et al. Neutralization of CXCL12 attenuates established pulmonary hypertension in rats. Cardiovasc. Res. 2020, 116, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, A.I.; Herbers, J.; Willenberg, I.; Chen, R.; Hwang, S.H.; Greite, R.; Morisseau, C.; Gueler, F.; Hammock, B.D.; Schebb, N.H. Oral treatment of rodents with soluble epoxide hydrolase inhibitor 1-(1-propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea (TPPU): Resulting drug levels and modulation of oxylipin pattern. Prostaglandins Other Lipid Mediat. 2015, 121, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, Y.; Phan, C.; Tu, L.; Le Hiress, M.; Thuillet, R.; Jutant, E.M.; Fadel, E.; Savale, L.; Huertas, A.; Humbert, M.; et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Investig. 2018, 128, 1956–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.; Desroches-Castan, A.; Mallet, C.; Guyon, L.; Cumont, A.; Phan, C.; Robert, F.; Thuillet, R.; Bordenave, J.; Sekine, A.; et al. Selective BMP-9 inhibition partially protects against experimental pulmonary hypertension. Circ. Res. 2019, 124, 846–855. [Google Scholar] [CrossRef]
- Feugray, G.; Pereira, T.; Iacob, M.; Moreau-Grangé, L.; Prévost, G.; Brunel, V.; Joannidès, R.; Bellien, J.; Duflot, T. Determination of Lipoxygenase, CYP450, and Non-Enzymatic Metabolites of Arachidonic Acid in Essential Hypertension and Type 2 Diabetes. Metabolites 2022, 12, 859. [Google Scholar] [CrossRef] [PubMed]
- Keserü, B.; Barbosa-Sicard, E.; Schermuly, R.T.; Tanaka, H.; Hammock, B.D.; Weissmann, N.; Fisslthaler, B.; Fleming, I. Hypoxia-induced pulmonary hypertension: Comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc. Res. 2010, 85, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Marowsky, A.; Meyer, I.; Erismann-Ebner, K.; Pellegrini, G.; Mule, N.; Arand, M. Beyond detoxification: A role for mouse mEH in the hepatic metabolism of endogenous lipids. Arch. Toxicol. 2017, 91, 3571–3585. [Google Scholar] [CrossRef] [Green Version]
- Edin, M.L.; Hamedani, B.G.; Gruzdev, A.; Graves, J.P.; Lih, F.B.; Arbes, S.J., 3rd; Singh, R.; Orjuela Leon, A.C.; Bradbury, J.A.; DeGraff, L.M.; et al. Epoxide hydrolase 1 (EPHX1) hydrolyzes epoxyeicosanoids and impairs cardiac recovery after ischemia. J. Biol. Chem. 2018, 293, 3281–3292. [Google Scholar] [CrossRef] [Green Version]
- Morisseau, C.; Kodani, S.D.; Kamita, S.G.; Yang, J.; Lee, K.S.S.; Hammock, B.D. Relative Importance of Soluble and Microsomal Epoxide Hydrolases for the Hydrolysis of Epoxy-Fatty Acids in Human Tissues. Int. J. Mol. Sci. 2021, 22, 4993. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, H.; Endo, J.; Kataoka, M.; Shimanaka, Y.; Kono, N.; Sugiura, Y.; Goto, S.; Kitakata, H.; Hiraide, T.; Yoshida, N.; et al. Omega-3 fatty acid epoxides produced by PAF-AH2 in mast cells regulate pulmonary vascular remodeling. Nat. Commun. 2022, 13, 3013. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.F. Cyclic AMPC-dependent modulation of cardiac L-type Ca2+ and transient outward K+ channel activities by epoxyeicosatrienoic acids. Prostaglandins Other Lipid Med. 2007, 81, 11–18. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leuillier, M.; Platel, V.; Tu, L.; Feugray, G.; Thuillet, R.; Groussard, D.; Messaoudi, H.; Ottaviani, M.; Chelgham, M.; Nicol, L.; et al. Inhibition of Soluble Epoxide Hydrolase Does Not Promote or Aggravate Pulmonary Hypertension in Rats. Cells 2023, 12, 665. https://doi.org/10.3390/cells12040665
Leuillier M, Platel V, Tu L, Feugray G, Thuillet R, Groussard D, Messaoudi H, Ottaviani M, Chelgham M, Nicol L, et al. Inhibition of Soluble Epoxide Hydrolase Does Not Promote or Aggravate Pulmonary Hypertension in Rats. Cells. 2023; 12(4):665. https://doi.org/10.3390/cells12040665
Chicago/Turabian StyleLeuillier, Matthieu, Valentin Platel, Ly Tu, Guillaume Feugray, Raphaël Thuillet, Déborah Groussard, Hind Messaoudi, Mina Ottaviani, Mustapha Chelgham, Lionel Nicol, and et al. 2023. "Inhibition of Soluble Epoxide Hydrolase Does Not Promote or Aggravate Pulmonary Hypertension in Rats" Cells 12, no. 4: 665. https://doi.org/10.3390/cells12040665