
Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity

 , , ,
, , ,  ,
,  , and
, and 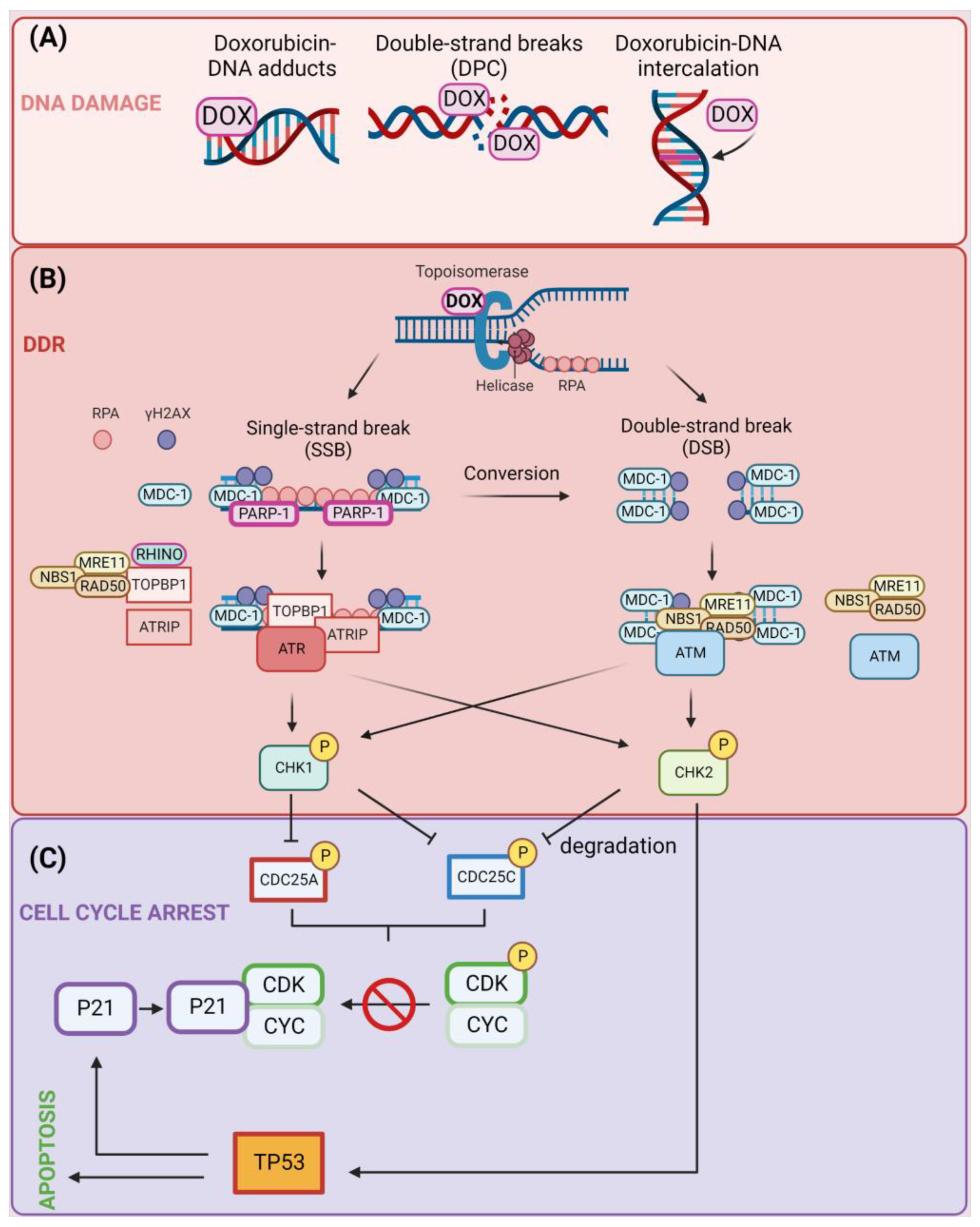{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mode of Action
2.1. Induction of DNA Damage
2.1.1. Formation of DOX–DNA Adducts and DNA Intercalation
2.1.2. Topoisomerase Trapping
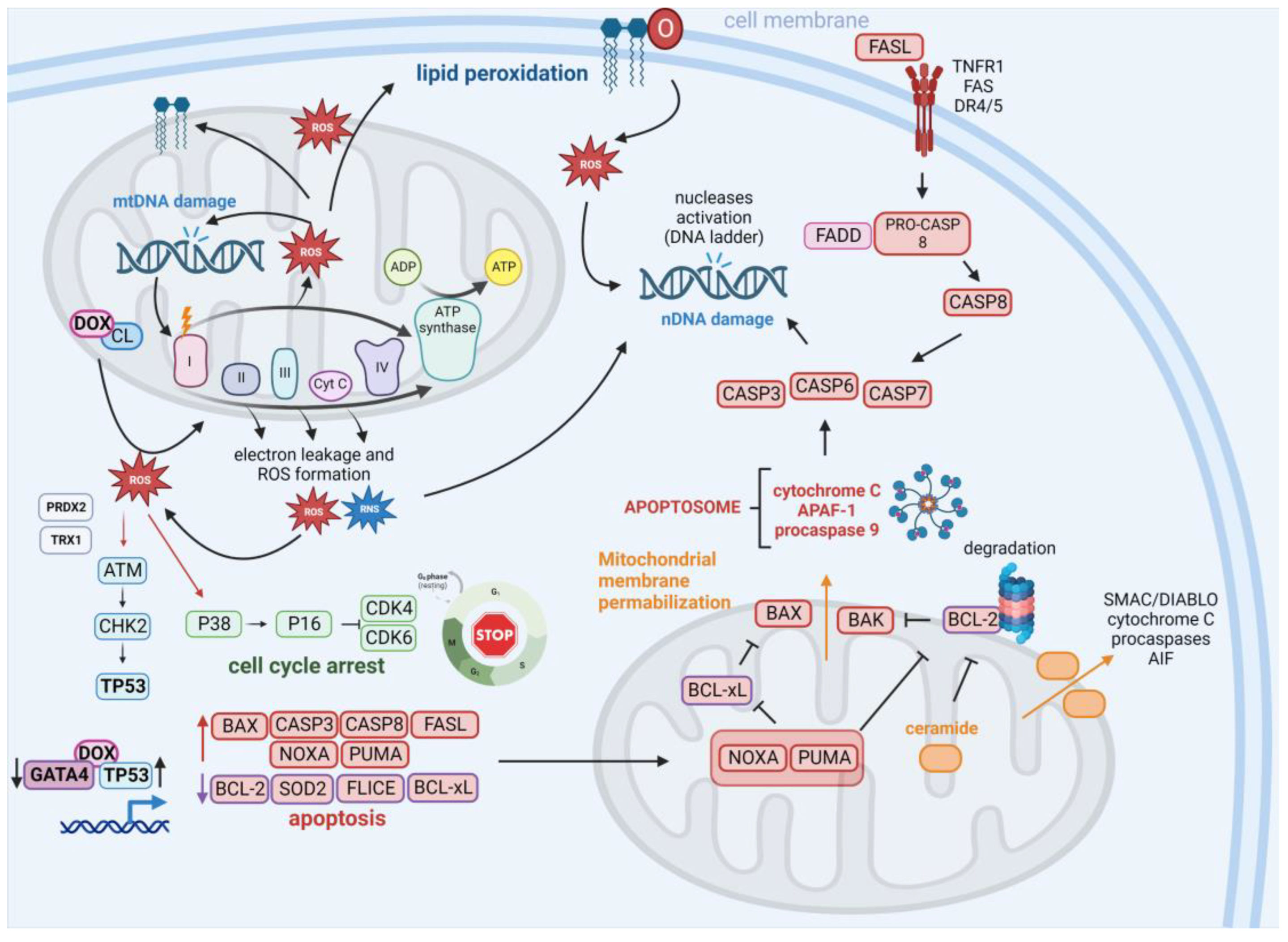
2.2. Apoptosis–ROS Interplay
2.3. Senescence Induction
2.4. Other Types of Cell Death
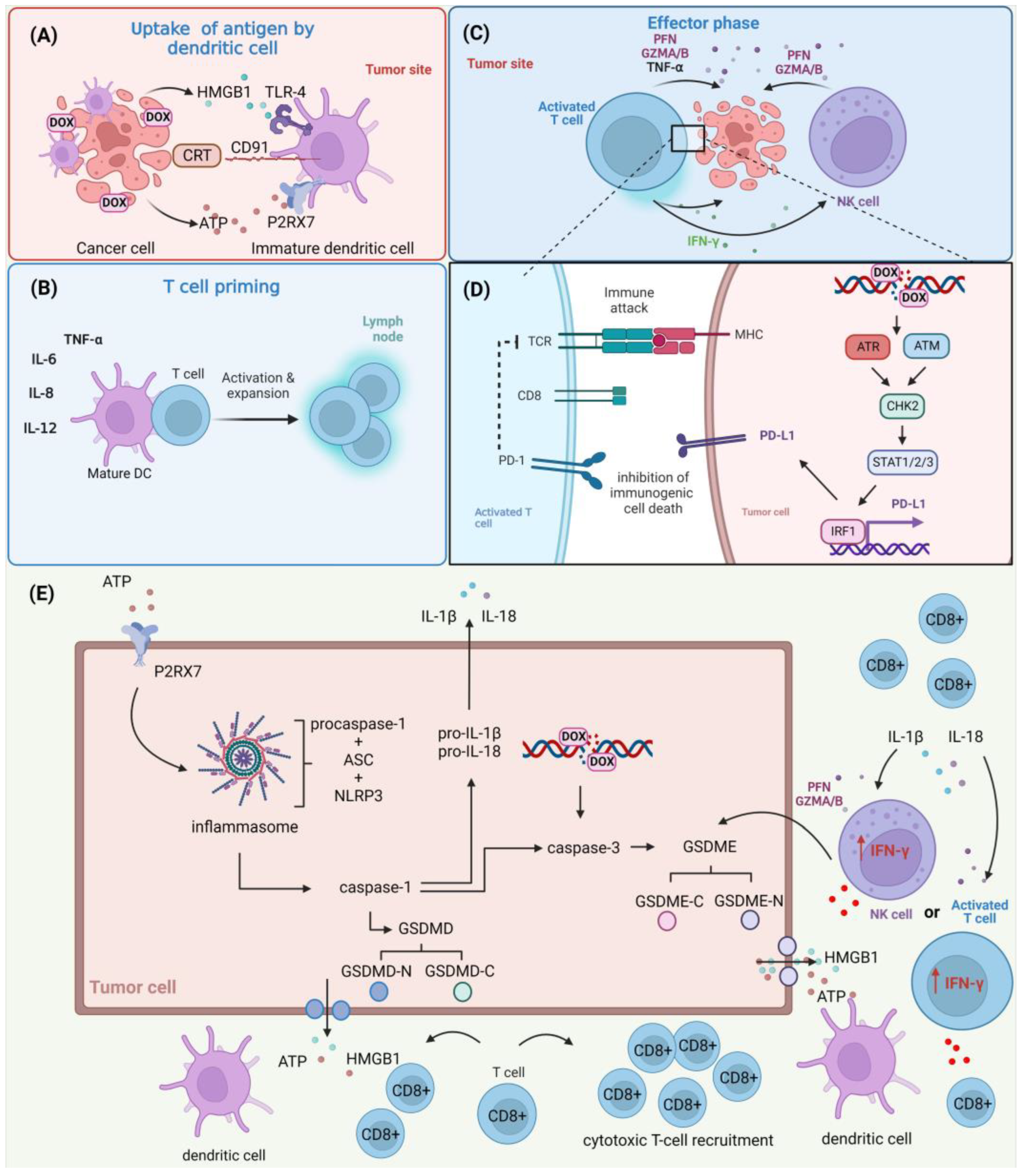
3. DOX and Tumor–Immune Microenvironment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| AIF | apoptosis-inducing factor |
| APAF1 | apoptotic protease-activating factor 1 |
| APC | antigen-presenting cell |
| ASC | adapter protein apoptosis-associated speck-like protein containing a CARD |
| ASCO | apoptosis-associated speck-like protein containing a caspase recruitment domain |
| ATM | ataxia telangiectasia mutated (A-T mutated) |
| ATR | ataxia telangiectasia and Rad3-related |
| ATRIP | ATR-interacting protein |
| BCL-2 | apoptosis regulator BCL2 |
| BCL-xL | B-cell lymphoma-extra large |
| CDC25A/C | M-phase inducer phosphatases |
| CDK | cyclin-dependent kinase |
| CHK1/2 | checkpoint kinases |
| CL | Cardiolipin |
| CRT | Calreticulin |
| CXCL | C-X-C motif chemokine receptors |
| DAMPs | damage-associated molecular patterns |
| DDR | DNA damage response |
| DFNA5 | deafness, autosomal dominant 5, isoform CRA |
| DIABLO | direct inhibitor of apoptosis-binding protein with low |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DOX | Doxorubicin |
| DPC | DNA–protein crosslink |
| DR5 | death receptor 5 |
| DSBs | double-strand breaks |
| eEF-2K | eukaryotic elongation factor-2 kinase |
| EMT | epithelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| ETAA1 | Ewing’s tumor-associated antigen 1 |
| ETC | electron transport chain |
| FASL | FAS antigen ligand |
| FDA | Food and Drug Administration |
| FLIP | FLICE/caspase-8 inhibitory protein |
| GATA4 | GATA Binding Protein 4 |
| GPX4 | glutathione peroxidase |
| GSDMD/E | gasdermin-D/E |
| GZMA/B | granzyme A/B |
| HMGB1 | high mobility group box protein B1 |
| HR | homologous recombination |
| IFNs | Interferons |
| ILs | Interleukins |
| IREs | iron-response elements |
| IRF1 | interferon regulatory factor 1 |
| IRP1/2 | iron regulatory proteins 1/2 |
| JAK | tyrosine-protein kinase JAK |
| JNK | C-Jun N-terminal kinase |
| MAVS | adaptor protein mitochondrial antiviral signaling |
| MCM2/7 | minichromosome maintenance helicase complex |
| MDA-5 | melanoma differentiation Ag-5 |
| MDC-1 | mediator of DNA damage checkpoint protein 1 |
| MDR1 | multidrug resistance 1 |
| MHC | major histocompatibility complex |
| MRE11 | meiotic recombination 11 homolog 1 |
| MRN | MRE11–RAD50–NBS1 complex |
| mtDNA | mitochondrial DNA |
| NBS1 | Nibrin |
| nDNA | nuclear DNA |
| NFAT | nuclear factor of activated T-lymphocytes |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHEJ | non-homologous end joining |
| NK | natural killer cell |
| NLRP3 | NACHT, LRR, and PYD domains-containing protein 3 |
| NOXA | phorbol-12-myristate-13-acetate-induced protein 1 |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| P2RX7 | P2X purinoceptor |
| PARP-1 | poly(ADP-ribose) polymerase 1 |
| PARs | poly(ADP-ribose) polymers |
| PD-L1 | programmed death ligand 1 |
| PEG | polyethylene glycol |
| PFNs | Perforins |
| PG-P | P-glycoprotein |
| PIKKs | phosphoinositide 3-kinase-related protein kinases |
| PNKP | polynucleotide kinase phosphatase |
| pRB | retinoblastoma protein |
| PRDX2 | peroxiredoxin-2 |
| PUMA | isoform 1 of Bcl-2-binding component 3, isoforms 1/2 |
| RHINO | RAD9, RAD1, HUS1 interacting nuclear orphan |
| RIG-1 | retinoic acid inducible gene-I |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RPA | replication protein A |
| SASP | senescence-associated secretory phenotype |
| SOD2 | superoxide dismutase 2 |
| SMAC | secondary mitochondria-derived activator of caspases |
| SSBR | single-strand break repair |
| SSBs | single-strand breaks |
| ssDNA | single-stranded DNA |
| STAT1/2/3 | signal transducer and activator of transcription 1/2/3 |
| TAMs | tumor-associated macrophages |
| TPD-1 | tyrosyl-DNA phosphodiesterase 1 |
| TIS | therapy-induced senescence |
| TLR4 | Toll-like receptor 4 |
| TNFR1 | tumor necrosis factor receptor 1 |
| TNF-α | tumor necrosis factor α |
| TOPB1 | topoisomerase 2-binding protein 1 |
| TP53 | cellular tumor antigen p53 |
| TRAIL | TNF-related apoptosis inducing ligand |
| TRX1 | thioredoxin-1 |
| XRCC1 | X-ray repair cross-complementing group-1 |
References
- Mohseni, M.; Samadi, N.; Ghanbari, P.; Yousefi, B.; Tabasinezhad, M.; Sharifi, S.; Nazemiyeh, H. Co-Treatment by Docetaxel and Vinblastine Breaks down P-Glycoprotein Mediated Chemo-Resistance. Iran. J. Basic Med. Sci. 2016, 19, 300–309. [Google Scholar]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of Conventional Anti-Cancer Drugs as New Tools against Multidrug Resistant Tumors. Drug Resist. Updat. 2020, 50, 100682. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-Induced Cardiotoxicity: An Update on the Molecular Mechanism and Novel Therapeutic Strategies for Effective Management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin Pathways: Pharmacodynamics and Adverse Effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Shi, Y.; Moon, M.; Dawood, S.; McManus, B.; Liu, P.P. Mechanisms and Management of Doxorubicin Cardiotoxicity. Herz 2011, 36, 296–305. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-Induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Sheibani, M.; Azizi, Y.; Shayan, M.; Nezamoleslami, S.; Eslami, F.; Farjoo, M.H.; Dehpour, A.R. Doxorubicin-Induced Cardiotoxicity: An Overview on Pre-Clinical Therapeutic Approaches. Cardiovasc. Toxicol. 2022, 22, 292–310. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the Basics of the Mechanism of Doxorubicin-Induced Cardiotoxicity: Have We Been Barking up the Wrong Tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Edison, T.N.J.I.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to Different Experimental Organ Systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Mohan, U.P.; Pichiah, P.B.T.; Iqbal, S.T.A.; Arunachalam, S. Mechanisms of Doxorubicin-Mediated Reproductive Toxicity—A Review. Reprod. Toxicol. 2021, 102, 80–89. [Google Scholar] [CrossRef]
- Alhowail, A.H.; Bloemer, J.; Majrashi, M.; Pinky, P.D.; Bhattacharya, S.; Yongli, Z.; Bhattacharya, D.; Eggert, M.; Woodie, L.; Buabeid, M.A.; et al. Doxorubicin-Induced Neurotoxicity Is Associated with Acute Alterations in Synaptic Plasticity, Apoptosis, and Lipid Peroxidation. Toxicol. Mech. Methods 2019, 29, 457–466. [Google Scholar] [CrossRef]
- Tangpong, J.; Miriyala, S.; Noel, T.; Sinthupibulyakit, C.; Jungsuwadee, P.; St Clair, D.K. Doxorubicin-Induced Central Nervous System Toxicity and Protection by Xanthone Derivative of Garcinia Mangostana. Neuroscience 2011, 175, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Prasanna, P.L.; Renu, K.; Valsala Gopalakrishnan, A. New Molecular and Biochemical Insights of Doxorubicin-Induced Hepatotoxicity. Life Sci. 2020, 250, 117599. [Google Scholar] [CrossRef]
- Ayla, S.; Seckin, I.; Tanriverdi, G.; Cengiz, M.; Eser, M.; Soner, B.C.; Oktem, G. Doxorubicin Induced Nephrotoxicity: Protective Effect of Nicotinamide. Int. J. Cell Biol. 2011, 2011, 390238. [Google Scholar] [CrossRef] [Green Version]
- Manil, L.; Couvreur, P.; Mahieu, P. Acute Renal Toxicity of Doxorubicin (Adriamycin)-Loaded Cyanoacrylate Nanoparticles. Pharm. Res. 1995, 12, 85–87. [Google Scholar] [CrossRef]
- Lahoti, T.S.; Patel, D.; Thekkemadom, V.; Beckett, R.; Ray, S.D. Doxorubicin-Induced in Vivo Nephrotoxicity Involves Oxidative Stress-Mediated Multiple pro- and Anti-Apoptotic Signaling Pathways. Curr. Neurovasc. Res. 2012, 9, 282–295. [Google Scholar] [CrossRef]
- van der Zanden, S.Y.; Qiao, X.; Neefjes, J. New Insights into the Activities and Toxicities of the Old Anticancer Drug Doxorubicin. FEBS J. 2021, 288, 6095–6111. [Google Scholar] [CrossRef]
- Varela-López, A.; Battino, M.; Navarro-Hortal, M.D.; Giampieri, F.; Forbes-Hernández, T.Y.; Romero-Márquez, J.M.; Collado, R.; Quiles, J.L. An Update on the Mechanisms Related to Cell Death and Toxicity of Doxorubicin and the Protective Role of Nutrients. Food Chem. Toxicol. 2019, 134, 110834. [Google Scholar] [CrossRef]
- Moustafa, I.; Viljoen, M.; Perumal-Pillay, V.A.; Oosthuizen, F. Critical Appraisal of Clinical Guidelines for Prevention and Management of Doxorubicin-Induced Cardiotoxicity. J. Oncol. Pharm. Pract. 2022, 147660. [Google Scholar] [CrossRef]
- Benzer, F.; Kandemir, F.M.; Kucukler, S.; Comaklı, S.; Caglayan, C. Chemoprotective Effects of Curcumin on Doxorubicin-Induced Nephrotoxicity in Wistar Rats: By Modulating Inflammatory Cytokines, Apoptosis, Oxidative Stress and Oxidative DNA Damage. Arch. Physiol. Biochem. 2018, 124, 448–457. [Google Scholar] [CrossRef]
- Rashid, S.; Ali, N.; Nafees, S.; Ahmad, S.T.; Arjumand, W.; Hasan, S.K.; Sultana, S. Alleviation of Doxorubicin-Induced Nephrotoxicity and Hepatotoxicity by Chrysin in Wistar Rats. Toxicol. Mech. Methods 2013, 23, 337–345. [Google Scholar] [CrossRef]
- Ibrahim Fouad, G.; Ahmed, K.A. Neuroprotective Potential of Berberine Against Doxorubicin-Induced Toxicity in Rat’s Brain. Neurochem. Res. 2021, 46, 3247–3263. [Google Scholar] [CrossRef]
- Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; El Habak, K.; Mostafa, R.; Ali, M.A.; et al. A Review on the Efficacy and Toxicity of Different Doxorubicin Nanoparticles for Targeted Therapy in Metastatic Breast Cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [Google Scholar] [CrossRef]
- Prados, J.; Melguizo, C.; Ortiz, R.; Vélez, C.; Alvarez, P.J.; Arias, J.L.; Ruíz, M.A.; Gallardo, V.; Aranega, A. Doxorubicin-Loaded Nanoparticles: New Advances in Breast Cancer Therapy. Anticancer Agents Med. Chem. 2012, 12, 1058–1070. [Google Scholar] [CrossRef]
- Zhao, N.; Woodle, M.C.; Mixson, A.J. Advances in Delivery Systems for Doxorubicin. J. Nanomed. Nanotechnol. 2018, 9, 519. [Google Scholar] [CrossRef]
- Kanwal, U.; Irfan Bukhari, N.; Ovais, M.; Abass, N.; Hussain, K.; Raza, A. Advances in Nano-Delivery Systems for Doxorubicin: An Updated Insight. J. Drug Target. 2018, 26, 296–310. [Google Scholar] [CrossRef]
- Ansari, L.; Shiehzadeh, F.; Taherzadeh, Z.; Nikoofal-Sahlabadi, S.; Momtazi-Borojeni, A.A.; Sahebkar, A.; Eslami, S. The Most Prevalent Side Effects of Pegylated Liposomal Doxorubicin Monotherapy in Women with Metastatic Breast Cancer: A Systematic Review of Clinical Trials. Cancer Gene Ther. 2017, 24, 189–193. [Google Scholar] [CrossRef]
- Miguel, R.D.A.; Hirata, A.S.; Jimenez, P.C.; Lopes, L.B.; Costa-Lotufo, L.V. Beyond Formulation: Contributions of Nanotechnology for Translation of Anticancer Natural Products into New Drugs. Pharmaceutics 2022, 14, 1722. [Google Scholar] [CrossRef]
- Mukhtar, M.; Bilal, M.; Rahdar, A.; Barani, M.; Arshad, R.; Behl, T.; Brisc, C.; Banica, F.; Bungau, S. Nanomaterials for Diagnosis and Treatment of Brain Cancer: Recent Updates. Chemosensors 2020, 8, 117. [Google Scholar] [CrossRef]
- Skarbek, C.; Serra, S.; Maslah, H.; Rascol, E.; Labruère, R. Arylboronate Prodrugs of Doxorubicin as Promising Chemotherapy for Pancreatic Cancer. Bioorg. Chem. 2019, 91, 103158. [Google Scholar] [CrossRef]
- Zhong, Y.-J.; Shao, L.-H.; Li, Y. Cathepsin B-Cleavable Doxorubicin Prodrugs for Targeted Cancer Therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Garsky, V.M.; Lumma, P.K.; Feng, D.M.; Wai, J.; Ramjit, H.G.; Sardana, M.K.; Oliff, A.; Jones, R.E.; DeFeo-Jones, D.; Freidinger, R.M. The Synthesis of a Prodrug of Doxorubicin Designed to Provide Reduced Systemic Toxicity and Greater Target Efficacy. J. Med. Chem. 2001, 44, 4216–4224. [Google Scholar] [CrossRef]
- DeFeo-Jones, D.; Garsky, V.M.; Wong, B.K.; Feng, D.M.; Bolyar, T.; Haskell, K.; Kiefer, D.M.; Leander, K.; McAvoy, E.; Lumma, P.; et al. A Peptide-Doxorubicin “prodrug” Activated by Prostate-Specific Antigen Selectively Kills Prostate Tumor Cells Positive for Prostate-Specific Antigen in Vivo. Nat. Med. 2000, 6, 1248–1252. [Google Scholar] [CrossRef]
- Albright, C.F.; Graciani, N.; Han, W.; Yue, E.; Stein, R.; Lai, Z.; Diamond, M.; Dowling, R.; Grimminger, L.; Zhang, S.-Y.; et al. Matrix Metalloproteinase-Activated Doxorubicin Prodrugs Inhibit HT1080 Xenograft Growth Better than Doxorubicin with Less Toxicity. Mol. Cancer Ther. 2005, 4, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Yin, M.; Hu, X.; Zhuang, X.; Sun, Y.; Guo, Y.; Tan, S.; Zhang, Z. A Safe, Simple and Efficient Doxorubicin Prodrug Hybrid Micelle for Overcoming Tumor Multidrug Resistance and Targeting Delivery. J. Control. Release 2016, 235, 182–194. [Google Scholar] [CrossRef]
- Farquhar, D.; Newman, R.A.; Zuckerman, J.E.; Andersson, B.S. Doxorubicin Analogues Incorporating Chemically Reactive Substituents. J. Med. Chem. 1991, 34, 561–564. [Google Scholar] [CrossRef]
- Weiss, R.B. The Anthracyclines: Will We Ever Find a Better Doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Brown, J.R.; Imam, S.H. Recent Studies on Doxorubicin and Its Analogues. Prog. Med. Chem. 1984, 21, 169–236. [Google Scholar] [CrossRef]
- Giuliani, F.C.; Kaplan, N.O. New Doxorubicin Analogs Active against Doxorubicin-Resistant Colon Tumor Xenografts in the Nude Mouse. Cancer Res. 1980, 40, 4682–4687. [Google Scholar]
- Lang, D.K.; Kaur, R.; Arora, R.; Saini, B.; Arora, S. Nitrogen-Containing Heterocycles as Anticancer Agents: An Overview. Anticancer Agents Med. Chem. 2020, 20, 2150–2168. [Google Scholar] [CrossRef]
- Sohail, M.; Sun, Z.; Li, Y.; Gu, X.; Xu, H. Research Progress in Strategies to Improve the Efficacy and Safety of Doxorubicin for Cancer Chemotherapy. Expert Rev. Anticancer Ther. 2021, 21, 1385–1398. [Google Scholar] [CrossRef]
- Zujewski, J.A.; Eng-Wong, J.; O’Shaughnessy, J.; Venzon, D.; Chow, C.; Danforth, D.; Kohler, D.R.; Cusack, G.; Riseberg, D.; Cowan, K.H. A Pilot Study of Dose Intense Doxorubicin and Cyclophosphamide Followed by Infusional Paclitaxel in High-Risk Primary Breast Cancer. Breast Cancer Res. Treat. 2003, 81, 41–51. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Wang, H.; Wu, X.; Yan, W.; Chen, Y.; Xu, Y.; Wang, C.; Yao, W.; Wang, J.; et al. Pegylated Liposomal Doxorubicin Combined with Ifosfamide for Treating Advanced or Metastatic Soft-Tissue Sarcoma: A Prospective, Single-Arm Phase II Study. Clin. Cancer Res. 2022, 28, 5280–5289. [Google Scholar] [CrossRef]
- Pautier, P.; Italiano, A.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Firmin, N.; Boudou-Rouquette, P.; Bertucci, F.; Balleyguier, C.; Lebrun-Ly, V.; et al. Doxorubicin Alone versus Doxorubicin with Trabectedin Followed by Trabectedin Alone as First-Line Therapy for Metastatic or Unresectable Leiomyosarcoma (LMS-04): A Randomised, Multicentre, Open-Label Phase 3 Trial. Lancet Oncol. 2022, 23, 1044–1054. [Google Scholar] [CrossRef]
- Picardi, M.; Giordano, C.; Pugliese, N.; Esposito, M.; Fatigati, M.; Muriano, F.; Rascato, M.G.; Pepa, R.D.; D’Ambrosio, A.; Vigliar, E.; et al. Liposomal Doxorubicin Supercharge-Containing Front-Line Treatment in Patients with Advanced-Stage Diffuse Large B-Cell Lymphoma or Classical Hodgkin Lymphoma: Preliminary Results of a Single-Centre Phase II Study. Br. J. Haematol. 2022, 198, 847–860. [Google Scholar] [CrossRef]
- Prakash, S.S.; Nagarkar, R.V.; Puligundla, K.C.; Lokesh, K.N.; Boya, R.R.; Patel, A.B.; Goyal, L.; Thoke, A.; Patel, J.G.; Mehta, A.O.; et al. Bioequivalence of a Hybrid Pegylated Liposomal Doxorubicin Hydrochloride Injection and Caelyx®: A Single-Dose, Randomized, Multicenter, Open-Label, Two-Period Crossover Study in Patients with Advanced Ovarian Cancer. Eur. J. Pharm. Sci. 2022, 176, 106248. [Google Scholar] [CrossRef]
- Wang, T.; Tang, J.; Yang, H.; Yin, R.; Zhang, J.; Zhou, Q.; Liu, Z.; Cao, L.; Li, L.; Huang, Y.; et al. Effect of Apatinib Plus Pegylated Liposomal Doxorubicin vs Pegylated Liposomal Doxorubicin Alone on Platinum-Resistant Recurrent Ovarian Cancer: The APPROVE Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1169–1176. [Google Scholar] [CrossRef]
- Lee, Y.J.; Seol, A.; Lee, M.; Kim, J.-W.; Kim, H.S.; Kim, K.; Suh, D.H.; Kim, S.; Kim, S.W.; Lee, J.-Y. A Phase II Trial to Evaluate the Efficacy of Bortezomib and Liposomal Doxorubicin in Patients With BRCA Wild-Type Platinum-Resistant Recurrent Ovarian Cancer (KGOG 3044/EBLIN). In Vivo 2022, 36, 1949–1958. [Google Scholar] [CrossRef]
- Elfadadny, A.; Ragab, R.F.; Hamada, R.; Al Jaouni, S.K.; Fu, J.; Mousa, S.A.; El-Far, A.H. Natural Bioactive Compounds-Doxorubicin Combinations Targeting Topoisomerase II-Alpha: Anticancer Efficacy and Safety. Toxicol. Appl. Pharmacol. 2023, 461, 116405. [Google Scholar] [CrossRef]
- Qiu, C.; Wu, Y.; Shi, Q.; Guo, Q.; Zhang, J.; Meng, Y.; Wang, C.; Xia, F.; Wang, J.; Xu, C. Advanced Strategies for Nucleic Acids and Small-Molecular Drugs in Combined Anticancer Therapy. Int. J. Biol. Sci. 2023, 19, 789–810. [Google Scholar] [CrossRef]
- de Padua, T.C.; Marandino, L.; Raggi, D.; Hallanger-Johnson, J.; Kutikov, A.; Spiess, P.E.; Necchi, A. A Systematic Review of Published Clinical Trials in the Systemic Treatment of Adrenocortical Carcinoma: An Initiative Led on Behalf of the Global Society of Rare Genitourinary Tumors. Clin. Genitourin. Cancer 2023, 21, 1–7. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, H.; Shan, N.; Qu, H. Pegylated Liposomal Doxorubicin (PLD)-Containing Regimen as a Novel Treatment of Monomorphic Epithelial Intestinal T-Cell Lymphoma (MEITL): A Case Report and Review of Literature. Medicine 2022, 101, e31326. [Google Scholar] [CrossRef]
- Younes, M.; Mardirossian, R.; Rizk, L.; Fazlian, T.; Khairallah, J.P.; Sleiman, C.; Naim, H.Y.; Rizk, S. The Synergistic Effects of Curcumin and Chemotherapeutic Drugs in Inhibiting Metastatic, Invasive and Proliferative Pathways. Plants 2022, 11, 2137. [Google Scholar] [CrossRef]
- Kciuk, M.; Kołat, D.; Kałuzińska-Kołat, Ż.; Gawrysiak, M.; Drozda, R.; Celik, I.; Kontek, R. PD-1/PD-L1 and DNA Damage Response in Cancer. Cells 2023, 12, 530. [Google Scholar] [CrossRef]
- Howerton, S.B.; Nagpal, A.; Williams, L.D. Surprising Roles of Electrostatic Interactions in DNA-Ligand Complexes. Biopolymers 2003, 69, 87–99. [Google Scholar] [CrossRef]
- Lipscomb, L.A.; Peek, M.E.; Zhou, F.X.; Bertrand, J.A.; VanDerveer, D.; Williams, L.D. Water Ring Structure at DNA Interfaces: Hydration and Dynamics of DNA-Anthracycline Complexes. Biochemistry 1994, 33, 3649–3659. [Google Scholar] [CrossRef]
- Zlatanova, J.; Victor, J.-M. How Are Nucleosomes Disrupted during Transcription Elongation? HFSP J. 2009, 3, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Kemp, C.J.; Henikoff, S. Doxorubicin Enhances Nucleosome Turnover around Promoters. Curr. Biol. 2013, 23, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Salerno, D.; Brogioli, D.; Cassina, V.; Turchi, D.; Beretta, G.L.; Seruggia, D.; Ziano, R.; Zunino, F.; Mantegazza, F. Magnetic Tweezers Measurements of the Nanomechanical Properties of DNA in the Presence of Drugs. Nucleic Acids Res. 2010, 38, 7089–7099. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA Torsion, and Chromatin Dynamics. Biochim. Biophys. Acta 2014, 1845, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Swift, L.P.; Rephaeli, A.; Nudelman, A.; Phillips, D.R.; Cutts, S.M. Doxorubicin-DNA Adducts Induce a Non-Topoisomerase II-Mediated Form of Cell Death. Cancer Res. 2006, 66, 4863–4871. [Google Scholar] [CrossRef] [Green Version]
- Forrest, R.A.; Swift, L.P.; Rephaeli, A.; Nudelman, A.; Kimura, K.-I.; Phillips, D.R.; Cutts, S.M. Activation of DNA Damage Response Pathways as a Consequence of Anthracycline-DNA Adduct Formation. Biochem. Pharmacol. 2012, 83, 1602–1612. [Google Scholar] [CrossRef]
- Coldwell, K.E.; Cutts, S.M.; Ognibene, T.J.; Henderson, P.T.; Phillips, D.R. Detection of Adriamycin-DNA Adducts by Accelerator Mass Spectrometry at Clinically Relevant Adriamycin Concentrations. Nucleic Acids Res. 2008, 36, e100. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan—Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Ross, W.E.; Glaubiger, D.L.; Kohn, K.W. Protein-Associated DNA Breaks in Cells Treated with Adriamycin or Ellipticine. Biochim. Biophys. Acta 1978, 519, 23–30. [Google Scholar] [CrossRef]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA Damage Response and Cancer Therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in Cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.; Dai, X. The Relationship between DNA Single-Stranded Damage Response and Double-Stranded Damage Response. Cell Cycle 2018, 17, 73–79. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Kołat, D.; Kałuzińska, Ż.; Kontek, R. Cancer-Associated Transcription Factors in DNA Damage Response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188757. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-Dependent Kinases in DNA Damage Response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Mojzych, M.; Kontek, R. Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies. Int. J. Mol. Sci. 2020, 21, 7264. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: An Essential Regulator of Genome Integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.; Lei, L.; Tan, L.-M.; Xu, X.-J.; He, B.-M.; Luo, C.; Yin, J.-Y.; Li, X.; Zhang, W.; Zhou, H.-H.; et al. The Role of Single Strand Break Repair Pathways in Cellular Responses to Camptothecin Induced DNA Damage. Biomed. Pharmacother. 2020, 125, 109875. [Google Scholar] [CrossRef]
- Abbotts, R.; Wilson, D.M. Coordination of DNA Single Strand Break Repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Fortini, P.; Dogliotti, E. Base Damage and Single-Strand Break Repair: Mechanisms and Functional Significance of Short- and Long-Patch Repair Subpathways. DNA Repair. 2007, 6, 398–409. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian Single-Strand Break Repair: Mechanisms and Links with Chromatin. DNA Repair. 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Spiegel, J.O.; Van Houten, B.; Durrant, J.D. PARP1: Structural Insights and Pharmacological Targets for Inhibition. DNA Repair. 2021, 103, 103125. [Google Scholar] [CrossRef]
- van den Bosch, M.; Bree, R.T.; Lowndes, N.F. The MRN Complex: Coordinating and Mediating the Response to Broken Chromosomes. EMBO Rep. 2003, 4, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Huang, J. MRN Complex Is an Essential Effector of DNA Damage Repair. J. Zhejiang Univ. Sci. B 2021, 22, 31–37. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN Complex in Double-Strand Break Repair and Telomere Maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.-K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Knudsen, K.E. DNA-PKcs: A Targetable Protumorigenic Protein Kinase. Cancer Res. 2022, 82, 523–533. [Google Scholar] [CrossRef]
- Amani, J.; Gorjizadeh, N.; Younesi, S.; Najafi, M.; Ashrafi, A.M.; Irian, S.; Gorjizadeh, N.; Azizian, K. Cyclin-Dependent Kinase Inhibitors (CDKIs) and the DNA Damage Response: The Link between Signaling Pathways and Cancer. DNA Repair. 2021, 102, 103103. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-Dependent Kinase Synthetic Lethality Partners in DNA Damage Response. Int. J. Mol. Sci. 2022, 23, 3555. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, Cyclins and CKIs: Roles beyond Cell Cycle Regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Kurz, E.U.; Douglas, P.; Lees-Miller, S.P. Doxorubicin Activates ATM-Dependent Phosphorylation of Multiple Downstream Targets in Part through the Generation of Reactive Oxygen Species. J. Biol. Chem. 2004, 279, 53272–53281. [Google Scholar] [CrossRef] [Green Version]
- Théard, D.; Coisy, M.; Ducommun, B.; Concannon, P.; Darbon, J.M. Etoposide and Adriamycin but Not Genistein Can Activate the Checkpoint Kinase Chk2 Independently of ATM/ATR. Biochem. Biophys. Res. Commun. 2001, 289, 1199–1204. [Google Scholar] [CrossRef]
- Ghelli Luserna Di Rorà, A.; Ghetti, M.; Ledda, L.; Ferrari, A.; Bocconcelli, M.; Padella, A.; Napolitano, R.; Fontana, M.C.; Liverani, C.; Imbrogno, E.; et al. Exploring the ATR-CHK1 Pathway in the Response of Doxorubicin-Induced DNA Damages in Acute Lymphoblastic Leukemia Cells. Cell Biol. Toxicol. 2021. [CrossRef]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.B.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical Evaluation of a Novel ATM Inhibitor, KU59403, in Vitro and in Vivo in P53 Functional and Dysfunctional Models of Human Cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Dean, D.; Hornicek, F.J.; Pollock, R.E.; Hoffman, R.M.; Duan, Z. ATR Inhibition Sensitizes Liposarcoma to Doxorubicin by Increasing DNA Damage. Am. J. Cancer Res. 2022, 12, 1577–1592. [Google Scholar]
- Baranski, Z.; Booij, T.H.; Cleton-Jansen, A.-M.; Price, L.S.; van de Water, B.; Bovée, J.V.M.G.; Hogendoorn, P.C.W.; Danen, E.H.J. Aven-Mediated Checkpoint Kinase Control Regulates Proliferation and Resistance to Chemotherapy in Conventional Osteosarcoma. J. Pathol. 2015, 236, 348–359. [Google Scholar] [CrossRef]
- Chung, S.W.; Kim, G.C.; Kweon, S.; Lee, H.; Choi, J.U.; Mahmud, F.; Chang, H.W.; Kim, J.W.; Son, W.-C.; Kim, S.Y.; et al. Metronomic Oral Doxorubicin in Combination of Chk1 Inhibitor MK-8776 for P53-Deficient Breast Cancer Treatment. Biomaterials 2018, 182, 35–43. [Google Scholar] [CrossRef]
- Weng, M.-T.; Tung, T.-H.; Lee, J.-H.; Wei, S.-C.; Lin, H.-L.; Huang, Y.-J.; Wong, J.-M.; Luo, J.; Sheu, J.-C. Enhancer of Rudimentary Homolog Regulates DNA Damage Response in Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 9357. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.; Grenier, J.; Martinez-Marignac, V.; Amrein, L.; Shawi, M.; Tokars, M.; Aloyz, R.; Panasci, L. Effects of the Novel DNA Dependent Protein Kinase Inhibitor, IC486241, on the DNA Damage Response to Doxorubicin and Cisplatin in Breast Cancer Cells. Investig. New Drugs 2012, 30, 1736–1742. [Google Scholar] [CrossRef]
- Park, H.J.; Bae, J.S.; Kim, K.M.; Moon, Y.J.; Park, S.-H.; Ha, S.H.; Hussein, U.K.; Zhang, Z.; Park, H.S.; Park, B.-H.; et al. The PARP Inhibitor Olaparib Potentiates the Effect of the DNA Damaging Agent Doxorubicin in Osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 107. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, Y.-S.; Kim, D.-K. Doxorubicin Exerts Cytotoxic Effects through Cell Cycle Arrest and Fas-Mediated Cell Death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef]
- Bar-On, O.; Shapira, M.; Hershko, D.D. Differential Effects of Doxorubicin Treatment on Cell Cycle Arrest and Skp2 Expression in Breast Cancer Cells. Anticancer Drugs 2007, 18, 1113–1121. [Google Scholar] [CrossRef]
- Ling, Y.H.; el-Naggar, A.K.; Priebe, W.; Perez-Soler, R. Cell Cycle-Dependent Cytotoxicity, G2/M Phase Arrest, and Disruption of P34cdc2/Cyclin B1 Activity Induced by Doxorubicin in Synchronized P388 Cells. Mol. Pharmacol. 1996, 49, 832–841. [Google Scholar]
- Lüpertz, R.; Wätjen, W.; Kahl, R.; Chovolou, Y. Dose- and Time-Dependent Effects of Doxorubicin on Cytotoxicity, Cell Cycle and Apoptotic Cell Death in Human Colon Cancer Cells. Toxicology 2010, 271, 115–121. [Google Scholar] [CrossRef]
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous Mechanisms of Reactive Oxygen Species (ROS) Generation. Postep. Hig. Med. Dosw. (Online) 2016, 70, 1150–1165. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively Generated Base Damage to Cellular DNA. Free Radic. Biol. Med. 2010, 49, 9–21. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A. Oxidative DNA Damage & Repair: An Introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of Reactive Oxygen Species (ROS) in Apoptosis Induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Kuczler, M.D.; Olseen, A.M.; Pienta, K.J.; Amend, S.R. ROS-Induced Cell Cycle Arrest as a Mechanism of Resistance in Polyaneuploid Cancer Cells (PACCs). Prog. Biophys. Mol. Biol. 2021, 165, 3–7. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Sengupta, S.; Bhattacharyya, A. Divergence to Apoptosis from ROS Induced Cell Cycle Arrest: Effect of Cadmium. Mutat. Res. 2009, 663, 22–31. [Google Scholar] [CrossRef]
- Victorelli, S.; Passos, J.F. Reactive Oxygen Species Detection in Senescent Cells. Methods Mol. Biol. 2019, 1896, 21–29. [Google Scholar] [CrossRef]
- Benkafadar, N.; François, F.; Affortit, C.; Casas, F.; Ceccato, J.-C.; Menardo, J.; Venail, F.; Malfroy-Camine, B.; Puel, J.-L.; Wang, J. ROS-Induced Activation of DNA Damage Responses Drives Senescence-Like State in Postmitotic Cochlear Cells: Implication for Hearing Preservation. Mol. Neurobiol. 2019, 56, 5950–5969. [Google Scholar] [CrossRef] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [Green Version]
- Bertram, C.; Hass, R. Cellular Responses to Reactive Oxygen Species-Induced DNA Damage and Aging. Biol. Chem. 2008, 389, 211–220. [Google Scholar] [CrossRef]
- Gilliam, L.A.A.; Moylan, J.S.; Patterson, E.W.; Smith, J.D.; Wilson, A.S.; Rabbani, Z.; Reid, M.B. Doxorubicin Acts via Mitochondrial ROS to Stimulate Catabolism in C2C12 Myotubes. Am. J. Physiol. Cell Physiol. 2012, 302, C195–C202. [Google Scholar] [CrossRef] [Green Version]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the Adriamycin-Cardiolipin Complex. Role in Mitochondrial Toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxidative Med. Cell. Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef] [Green Version]
- Montalvo, R.N.; Doerr, V.; Min, K.; Szeto, H.H.; Smuder, A.J. Doxorubicin-Induced Oxidative Stress Differentially Regulates Proteolytic Signaling in Cardiac and Skeletal Muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R227–R233. [Google Scholar] [CrossRef]
- Kalivendi, S.V.; Konorev, E.A.; Cunningham, S.; Vanamala, S.K.; Kaji, E.H.; Joseph, J.; Kalyanaraman, B. Doxorubicin Activates Nuclear Factor of Activated T-Lymphocytes and Fas Ligand Transcription: Role of Mitochondrial Reactive Oxygen Species and Calcium. Biochem. J. 2005, 389, 527–539. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin Induces Cardiotoxicity through Upregulation of Death Receptors Mediated Apoptosis in Cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [Green Version]
- McSweeney, K.M.; Bozza, W.P.; Alterovitz, W.-L.; Zhang, B. Transcriptomic Profiling Reveals P53 as a Key Regulator of Doxorubicin-Induced Cardiotoxicity. Cell Death Discov. 2019, 5, 102. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Palmeira, C.M.; Wallace, K.B. Doxorubicin-Induced Persistent Oxidative Stress to Cardiac Myocytes. Toxicol. Lett. 2001, 121, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the Molecular Basis of Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Xie, R.; Liu, R.; Lu, Y. Mitochondria-Derived Reactive Oxygen Species Play an Important Role in Doxorubicin-Induced Platelet Apoptosis. Int. J. Mol. Sci. 2015, 16, 11087–11100. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Kim, S.-J.; Kim, B.-J.; Rah, S.-Y.; Chung, S.M.; Im, M.-J.; Kim, U.-H. Doxorubicin-Induced Reactive Oxygen Species Generation and Intracellular Ca2+ Increase Are Reciprocally Modulated in Rat Cardiomyocytes. Exp. Mol. Med. 2006, 38, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wang, Y.; Zheng, D.; Wei, M.; Xu, H.; Peng, T. Rac1 Signalling Mediates Doxorubicin-Induced Cardiotoxicity through Both Reactive Oxygen Species-Dependent and -Independent Pathways. Cardiovasc. Res. 2013, 97, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Asensio-López, M.C.; Soler, F.; Pascual-Figal, D.; Fernández-Belda, F.; Lax, A. Doxorubicin-Induced Oxidative Stress: The Protective Effect of Nicorandil on HL-1 Cardiomyocytes. PLoS ONE 2017, 12, e0172803. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabò, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The Determining Role of Mitochondrial Reactive Oxygen Species Generation and Monoamine Oxidase Activity in Doxorubicin-Induced Cardiotoxicity. Antioxid. Redox Signal. 2021, 34, 531–550. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative Stress Injury in Doxorubicin-Induced Cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Olson, R.D.; Mushlin, P.S. Doxorubicin Cardiotoxicity: Analysis of Prevailing Hypotheses. FASEB J. 1990, 4, 3076–3086. [Google Scholar] [CrossRef]
- Dos Santos Arruda, F.; Tomé, F.D.; Miguel, M.P.; de Menezes, L.B.; Nagib, P.R.A.; Campos, E.C.; Soave, D.F.; Celes, M.R.N. Doxorubicin-Induced Cardiotoxicity and Cardioprotective Agents: Classic and New Players in the Game. Curr. Pharm. Des. 2019, 25, 109–118. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxidative Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Zhang, Y.; Wang, Z.; Wang, S.; Jiang, X.; Cui, H.; Zhou, T.; He, Z.; Feng, H.; Guo, Q.; et al. ATM at the Crossroads of Reactive Oxygen Species and Autophagy. Int. J. Biol. Sci. 2021, 17, 3080–3090. [Google Scholar] [CrossRef]
- Sarkar, A.; Gandhi, V. Activation of ATM Kinase by ROS Generated during Ionophore-Induced Mitophagy in Human T and B Cell Malignancies. Mol. Cell. Biochem. 2021, 476, 417–423. [Google Scholar] [CrossRef]
- Tsang, W.P.; Chau, S.P.Y.; Kong, S.K.; Fung, K.P.; Kwok, T.T. Reactive Oxygen Species Mediate Doxorubicin Induced P53-Independent Apoptosis. Life Sci. 2003, 73, 2047–2058. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of Doxorubicin on Apoptosis and Oxidative Stress in Breast Cancer Cell Lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Navarro, R.; Busnadiego, I.; Ruiz-Larrea, M.B.; Ruiz-Sanz, J.I. Superoxide Anions Are Involved in Doxorubicin-Induced ERK Activation in Hepatocyte Cultures. Ann. N. Y. Acad. Sci. 2006, 1090, 419–428. [Google Scholar] [CrossRef]
- Filippova, M.; Filippov, V.; Williams, V.M.; Zhang, K.; Kokoza, A.; Bashkirova, S.; Duerksen-Hughes, P. Cellular Levels of Oxidative Stress Affect the Response of Cervical Cancer Cells to Chemotherapeutic Agents. Biomed. Res. Int. 2014, 2014, 574659. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Chanvorachote, P.; Nimmannit, U.; Leonard, S.S.; Stehlik, C.; Wang, L.; Rojanasakul, Y. Mitochondrial Superoxide Mediates Doxorubicin-Induced Keratinocyte Apoptosis through Oxidative Modification of ERK and Bcl-2 Ubiquitination. Biochem. Pharmacol. 2012, 83, 1643–1654. [Google Scholar] [CrossRef] [Green Version]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential Role of GATA-4 in Cell Survival and Drug-Induced Cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, J.H. Mechanisms of Anthracycline-Enhanced Reactive Oxygen Metabolism in Tumor Cells. Oxidative Med. Cell. Longev. 2019, 2019, 9474823. [Google Scholar] [CrossRef] [Green Version]
- Voulgaridou, G.-P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA Damage Induced by Endogenous Aldehydes: Current State of Knowledge. Mutat. Res. 2011, 711, 13–27. [Google Scholar] [CrossRef]
- Antonowicz, S.; Bodai, Z.; Wiggins, T.; Markar, S.R.; Boshier, P.R.; Goh, Y.M.; Adam, M.E.; Lu, H.; Kudo, H.; Rosini, F.; et al. Endogenous Aldehyde Accumulation Generates Genotoxicity and Exhaled Biomarkers in Esophageal Adenocarcinoma. Nat. Commun. 2021, 12, 1454. [Google Scholar] [CrossRef]
- Nakamura, J.; Nakamura, M. DNA-Protein Crosslink Formation by Endogenous Aldehydes and AP Sites. DNA Repair. 2020, 88, 102806. [Google Scholar] [CrossRef]
- Hlaváčová, M.; Gumulec, J.; Stračina, T.; Fojtů, M.; Raudenská, M.; Masařík, M.; Nováková, M.; Paulová, H. Different Doxorubicin Formulations Affect Plasma 4-Hydroxy-2-Nonenal and Gene Expression of Aldehyde Dehydrogenase 3A1 and Thioredoxin Reductase 2 in Rat. Physiol. Res. 2015, 64, S653–S660. [Google Scholar] [CrossRef]
- Luo, X.; Evrovsky, Y.; Cole, D.; Trines, J.; Benson, L.N.; Lehotay, D.C. Doxorubicin-Induced Acute Changes in Cytotoxic Aldehydes, Antioxidant Status and Cardiac Function in the Rat. Biochim. Biophys. Acta 1997, 1360, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Hrelia, S.; Fiorentini, D.; Maraldi, T.; Angeloni, C.; Bordoni, A.; Biagi, P.L.; Hakim, G. Doxorubicin Induces Early Lipid Peroxidation Associated with Changes in Glucose Transport in Cultured Cardiomyocytes. Biochim. Biophys. Acta 2002, 1567, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting Ceramide Metabolism--a Strategy for Overcoming Drug Resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef]
- Kawase, M.; Watanabe, M.; Kondo, T.; Yabu, T.; Taguchi, Y.; Umehara, H.; Uchiyama, T.; Mizuno, K.; Okazaki, T. Increase of Ceramide in Adriamycin-Induced HL-60 Cell Apoptosis: Detection by a Novel Anti-Ceramide Antibody. Biochim. Biophys. Acta 2002, 1584, 104–114. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Synold, T.W.; Somlo, G.; Akman, S.A.; Gajewski, E. Oxidative DNA Base Modifications in Peripheral Blood Mononuclear Cells of Patients Treated with High-Dose Infusional Doxorubicin. Blood 2001, 97, 2839–2845. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, E.; Gaur, S.; Akman, S.A.; Matsumoto, L.; van Balgooy, J.N.A.; Doroshow, J.H. Oxidative DNA Base Damage in MCF-10A Breast Epithelial Cells at Clinically Achievable Concentrations of Doxorubicin. Biochem. Pharmacol. 2007, 73, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct Effect of Ceramide on the Mitochondrial Electron Transport Chain Leads to Generation of Reactive Oxygen Species. Role of Mitochondrial Glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Fong, Y.; Tsai, E.-M.; Chang, Y.-G.; Chou, H.L.; Wu, C.-Y.; Teng, Y.-N.; Liu, T.-C.; Yuan, S.-S.; Chiu, C.-C. Exogenous C8-Ceramide Induces Apoptosis by Overproduction of ROS and the Switch of Superoxide Dismutases SOD1 to SOD2 in Human Lung Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3010. [Google Scholar] [CrossRef] [Green Version]
- Siskind, L.J. Mitochondrial Ceramide and the Induction of Apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef]
- Woodcock, J. Sphingosine and Ceramide Signalling in Apoptosis. IUBMB Life 2006, 58, 462–466. [Google Scholar] [CrossRef]
- Mullen, T.D.; Obeid, L.M. Ceramide and Apoptosis: Exploring the Enigmatic Connections between Sphingolipid Metabolism and Programmed Cell Death. Anticancer Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef]
- Khodadust, R.; Alpsoy, A.; Ünsoy, G.; GÜndÜz, U. Poly (I:C)- and Doxorubicin-Loaded Magnetic Dendrimeric Nanoparticles Affect the Apoptosis-Related Gene Expressions in MCF-7 Cells. Turk. J. Biol. 2020, 44, 133–144. [Google Scholar] [CrossRef]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, H. Doxorubicin-Induced Cancer Cell Senescence Shows a Time Delay Effect and Is Inhibited by Epithelial-Mesenchymal Transition (EMT). Med. Sci. Monit. 2019, 25, 3617–3623. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell. Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef]
- Joyner, D.E.; Bastar, J.D.; Randall, R.L. Doxorubicin Induces Cell Senescence Preferentially over Apoptosis in the FU-SY-1 Synovial Sarcoma Cell Line. J. Orthop. Res. 2006, 24, 1163–1169. [Google Scholar] [CrossRef]
- Strzeszewska, A.; Alster, O.; Mosieniak, G.; Ciolko, A.; Sikora, E. Insight into the Role of PIKK Family Members and NF-KB in DNAdamage-Induced Senescence and Senescence-Associated Secretory Phenotype of Colon Cancer Cells. Cell Death Dis. 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Roninson, I.B. Tumor Cell Senescence in Cancer Treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Yang, L.; Fang, J.; Chen, J. Tumor Cell Senescence Response Produces Aggressive Variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [Green Version]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of Senescence with Doxorubicin Leads to Increased Genomic Instability of HCT116 Cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef]
- Karabicici, M.; Alptekin, S.; Fırtına Karagonlar, Z.; Erdal, E. Doxorubicin-Induced Senescence Promotes Stemness and Tumorigenicity in EpCAM-/CD133- Nonstem Cell Population in Hepatocellular Carcinoma Cell Line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef]
- Yang, M.-Y.; Lin, P.-M.; Liu, Y.-C.; Hsiao, H.-H.; Yang, W.-C.; Hsu, J.-F.; Hsu, C.-M.; Lin, S.-F. Induction of Cellular Senescence by Doxorubicin Is Associated with Upregulated MiR-375 and Induction of Autophagy in K562 Cells. PLoS ONE 2012, 7, e37205. [Google Scholar] [CrossRef] [Green Version]
- Bientinesi, E.; Lulli, M.; Becatti, M.; Ristori, S.; Margheri, F.; Monti, D. Doxorubicin-Induced Senescence in Normal Fibroblasts Promotes in Vitro Tumour Cell Growth and Invasiveness: The Role of Quercetin in Modulating These Processes. Mech. Ageing Dev. 2022, 206, 111689. [Google Scholar] [CrossRef]
- Marques, L.; Johnson, A.A.; Stolzing, A. Doxorubicin Generates Senescent Microglia That Exhibit Altered Proteomes, Higher Levels of Cytokine Secretion, and a Decreased Ability to Internalize Amyloid β. Exp. Cell Res. 2020, 395, 112203. [Google Scholar] [CrossRef]
- Piegari, E.; De Angelis, A.; Cappetta, D.; Russo, R.; Esposito, G.; Costantino, S.; Graiani, G.; Frati, C.; Prezioso, L.; Berrino, L.; et al. Doxorubicin Induces Senescence and Impairs Function of Human Cardiac Progenitor Cells. Basic Res. Cardiol. 2013, 108, 334. [Google Scholar] [CrossRef]
- Spallarossa, P.; Altieri, P.; Aloi, C.; Garibaldi, S.; Barisione, C.; Ghigliotti, G.; Fugazza, G.; Barsotti, A.; Brunelli, C. Doxorubicin Induces Senescence or Apoptosis in Rat Neonatal Cardiomyocytes by Regulating the Expression Levels of the Telomere Binding Factors 1 and 2. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2169–H2181. [Google Scholar] [CrossRef] [Green Version]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated Cardiomyocyte Senescence Contributes to Late-Onset Doxorubicin-Induced Cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, C380–C391. [Google Scholar] [CrossRef]
- Bashiri Dezfouli, A.; Salar-Amoli, J.; Pourfathollah, A.A.; Yazdi, M.; Nikougoftar-Zarif, M.; Khosravi, M.; Hassan, J. Doxorubicin-Induced Senescence through NF-ΚB Affected by the Age of Mouse Mesenchymal Stem Cells. J. Cell. Physiol. 2020, 235, 2336–2349. [Google Scholar] [CrossRef]
- Sultana, R.; Di Domenico, F.; Tseng, M.; Cai, J.; Noel, T.; Chelvarajan, R.L.; Pierce, W.D.; Cini, C.; Bondada, S.; St Clair, D.K.; et al. Doxorubicin-Induced Thymus Senescence. J. Proteome Res. 2010, 9, 6232–6241. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of Autophagy in Cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Zhao, D.; Yuan, H.; Yi, F.; Meng, C.; Zhu, Q. Autophagy Prevents Doxorubicin-induced Apoptosis in Osteosarcoma. Mol. Med. Rep. 2014, 9, 1975–1981. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Tam, A.; Santi, S.A.; Parissenti, A.M. Role of Autophagy and Lysosomal Drug Sequestration in Acquired Resistance to Doxorubicin in MCF-7 Cells. BMC Cancer 2016, 16, 762. [Google Scholar] [CrossRef] [Green Version]
- Sishi, B.J.N.; Loos, B.; van Rooyen, J.; Engelbrecht, A.-M. Autophagy Upregulation Promotes Survival and Attenuates Doxorubicin-Induced Cardiotoxicity. Biochem. Pharmacol. 2013, 85, 124–134. [Google Scholar] [CrossRef]
- Loh, J.S.; Rahim, N.A.; Tor, Y.S.; Foo, J.B. Simultaneous Proteasome and Autophagy Inhibition Synergistically Enhances Cytotoxicity of Doxorubicin in Breast Cancer Cells. Cell Biochem. Funct. 2022, 40, 403–416. [Google Scholar] [CrossRef]
- Aydinlik, S.; Erkisa, M.; Cevatemre, B.; Sarimahmut, M.; Dere, E.; Ari, F.; Ulukaya, E. Enhanced Cytotoxic Activity of Doxorubicin through the Inhibition of Autophagy in Triple Negative Breast Cancer Cell Line. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 49–57. [Google Scholar] [CrossRef]
- Hu, C.; Gu, F.; Gong, C.; Xia, Q.; Gao, Y.; Gao, S. Co-Delivery of the Autophagy Inhibitor Si-Beclin1 and the Doxorubicin Nano-Delivery System for Advanced Prostate Cancer Treatment. J. Biomater. Appl. 2022, 36, 1317–1331. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Yang, R.; Li, Y.; Wang, X.; Yan, J.; Pan, D.; Xu, Y.; Wang, L.; Yang, M. Doxorubicin Loaded Ferritin Nanoparticles for Ferroptosis Enhanced Targeted Killing of Cancer Cells. RSC Adv. 2019, 9, 28548–28553. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.-C.; Li, M.-H.; Zhao, Y.; Zhou, J.; Hu, Y.; Cai, K.-Y.; Zhao, Y.; Yu, S.-H.; Luo, Z. Tumor Microenvironment-Activatable Fe-Doxorubicin Preloaded Amorphous CaCO3 Nanoformulation Triggers Ferroptosis in Target Tumor Cells. Sci. Adv. 2020, 6, eaax1346. [Google Scholar] [CrossRef]
- Yang, Y.; Zuo, S.; Li, L.; Kuang, X.; Li, J.; Sun, B.; Wang, S.; He, Z.; Sun, J. Iron-Doxorubicin Prodrug Loaded Liposome Nanogenerator Programs Multimodal Ferroptosis for Efficient Cancer Therapy. Asian J. Pharm. Sci. 2021, 16, 784–793. [Google Scholar] [CrossRef]
- Ji, P.; Wang, X.; Yin, J.; Yao, Y.; Du, W. Amplification of Ferroptosis with a Liposomal Nanoreactor Cooperates with Low-Toxicity Doxorubicin Apoptosis for Enhanced Tumor Chemotherapy. Biomater. Sci. 2022, 10, 1544–1553. [Google Scholar] [CrossRef]
- Bano, I.; Horky, P.; Abbas, S.Q.; Majid, M.; Bilal, A.H.M.; Ali, F.; Behl, T.; Shams ul Hassan, S.; Bungau, S. Ferroptosis: A New Road towards Cancer Management. Molecules 2022, 27, 2129. [Google Scholar] [CrossRef]
- Wang, L.; Qin, X.; Liang, J.; Ge, P. Induction of Pyroptosis: A Promising Strategy for Cancer Treatment. Front. Oncol. 2021, 11, 635774. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The Role of Pyroptosis in Cancer: Pro-Cancer or pro-“Host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Lin, H.; Zhang, J.; Lin, N.; Sun, Z.; Gao, F.; Luo, H.; Ni, T.; Luo, W.; Chi, J.; et al. Doxorubicin Induces Cardiomyocyte Pyroptosis via the TINCR-Mediated Posttranscriptional Stabilization of NLR Family Pyrin Domain Containing 3. J. Mol. Cell. Cardiol. 2019, 136, 15–26. [Google Scholar] [CrossRef]
- Dessouki, F.B.A.; Kukreja, R.C.; Singla, D.K. Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Muscle Toxicity through Counteracting Pyroptosis. Pharmaceuticals 2020, 13, 450. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Li, D.; Zhou, X.; Qin, Q.; Zhang, Q. Caspase-3-Mediated GSDME Induced Pyroptosis in Breast Cancer Cells through the ROS/JNK Signalling Pathway. J. Cell. Mol. Med. 2021, 25, 8159–8168. [Google Scholar] [CrossRef]
- Yu, P.; Wang, H.-Y.; Tian, M.; Li, A.-X.; Chen, X.-S.; Wang, X.-L.; Zhang, Y.; Cheng, Y. Eukaryotic Elongation Factor-2 Kinase Regulates the Cross-Talk between Autophagy and Pyroptosis in Doxorubicin-Treated Human Melanoma Cells in Vitro. Acta Pharmacol. Sin. 2019, 40, 1237–1244. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Zarganes-Tzitzikas, T.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies? Angew. Chem. Int. Ed. Engl. 2018, 57, 4840–4848. [Google Scholar] [CrossRef]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer Metal Drugs and Immunogenic Cell Death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 71, 189–194. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Mattarollo, S.R.; Loi, S.; Duret, H.; Ma, Y.; Zitvogel, L.; Smyth, M.J. Pivotal Role of Innate and Adaptive Immunity in Anthracycline Chemotherapy of Established Tumors. Cancer Res. 2011, 71, 4809–4820. [Google Scholar] [CrossRef] [Green Version]
- Park, J.Y.; Jang, M.J.; Chung, Y.H.; Kim, K.Y.; Kim, S.S.; Lee, W.B.; You, S.; Choi, Y.S.; Hur, D.Y.; Kim, D. Doxorubicin Enhances CD4(+) T-Cell Immune Responses by Inducing Expression of CD40 Ligand and 4-1BB. Int. Immunopharmacol. 2009, 9, 1530–1539. [Google Scholar] [CrossRef]
- Zirakzadeh, A.A.; Kinn, J.; Krantz, D.; Rosenblatt, R.; Winerdal, M.E.; Hu, J.; Hartana, C.A.; Lundgren, C.; Bergman, E.A.; Johansson, M.; et al. Doxorubicin Enhances the Capacity of B Cells to Activate T Cells in Urothelial Urinary Bladder Cancer. Clin. Immunol. 2017, 176, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Bedi, D.; Henderson, H.J.; Manne, U.; Samuel, T. Camptothecin Induces PD-L1 and Immunomodulatory Cytokines in Colon Cancer Cells. Medicines 2019, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Gilad, Y.; Eliaz, Y.; Yu, Y.; Han, S.J.; O’Malley, B.W.; Lonard, D.M. Drug-Induced PD-L1 Expression and Cell Stress Response in Breast Cancer Cells Can Be Balanced by Drug Combination. Sci. Rep. 2019, 9, 15099. [Google Scholar] [CrossRef] [Green Version]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune Induction Strategies in Metastatic Triple-Negative Breast Cancer to Enhance the Sensitivity to PD-1 Blockade: The TONIC Trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Wang, J.; Hu, C.; Wang, J.; Shen, Y.; Bao, Q.; He, F.; Wang, H.; Gong, L.; Liu, Z.; Hu, F.; et al. Checkpoint Blockade in Combination With Doxorubicin Augments Tumor Cell Apoptosis in Osteosarcoma. J. Immunother. 2019, 42, 321–330. [Google Scholar] [CrossRef]
- Iwai, T.; Sugimoto, M.; Wakita, D.; Yorozu, K.; Kurasawa, M.; Yamamoto, K. Topoisomerase I Inhibitor, Irinotecan, Depletes Regulatory T Cells and up-Regulates MHC Class I and PD-L1 Expression, Resulting in a Supra-Additive Antitumor Effect When Combined with Anti-PD-L1 Antibodies. Oncotarget 2018, 9, 31411–31421. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Xu, J.; Cai, H.; Lang, J. Carboplatin and Programmed Death-Ligand 1 Blockade Synergistically Produce a Similar Antitumor Effect to Carboplatin Alone in Murine ID8 Ovarian Cancer Model. J. Obstet. Gynaecol. Res. 2018, 44, 303–311. [Google Scholar] [CrossRef]
- Wahba, J.; Natoli, M.; Whilding, L.M.; Parente-Pereira, A.C.; Jung, Y.; Zona, S.; Lam, E.W.-F.; Smith, J.R.; Maher, J.; Ghaem-Maghami, S. Chemotherapy-Induced Apoptosis, Autophagy and Cell Cycle Arrest Are Key Drivers of Synergy in Chemo-Immunotherapy of Epithelial Ovarian Cancer. Cancer Immunol. Immunother. 2018, 67, 1753–1765. [Google Scholar] [CrossRef] [Green Version]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Ségal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Trédaniel, J.; et al. Cisplatin Increases PD-L1 Expression and Optimizes Immune Check-Point Blockade in Non-Small Cell Lung Cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.-J.; Spielbauer, K.; Van Waes, C.; Schmitt, N.C. Cisplatin Alters Antitumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Ock, C.-Y.; Kim, S.; Keam, B.; Kim, S.; Ahn, Y.-O.; Chung, E.-J.; Kim, J.-H.; Kim, T.M.; Kwon, S.K.; Jeon, Y.K.; et al. Changes in Programmed Death-Ligand 1 Expression during Cisplatin Treatment in Patients with Head and Neck Squamous Cell Carcinoma. Oncotarget 2017, 8, 97920–97927. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.-F.; Lin, J.-F.; Lin, Y.-C.; Chou, K.-Y.; Chen, H.-E.; Ho, C.-Y.; Chen, P.-C.; Hwang, T.I.-S. Cisplatin Contributes to Programmed Death-Ligand 1 Expression in Bladder Cancer through ERK1/2-AP-1 Signaling Pathway. Biosci. Rep. 2019, 39, BSR20190362. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Liu, C.; Zhou, Y.; Wang, G. Cisplatin Induces Programmed Death-1-Ligand 1(PD-L1) over-Expression in Hepatoma H22 Cells via Erk /MAPK Signaling Pathway. Cell. Mol. Biol. 2010, 56, 1366–1372. [Google Scholar]
- Wu, X.; Li, Y.; Liu, X.; Chen, C.; Harrington, S.M.; Cao, S.; Xie, T.; Pham, T.; Mansfield, A.S.; Yan, Y.; et al. Targeting B7-H1 (PD-L1) Sensitizes Cancer Cells to Chemotherapy. Heliyon 2018, 4, e01039. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT Pathway Is Involved in the Upregulation of PD-L1 Expression in Pancreatic Cancer Cell Lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Hu, J.; Hu, X.; Chen, H.; Bao, R.; Zhou, Y.; Ye, Y.; Zhan, M.; Cai, W.; Li, H.; et al. DENR Controls JAK2 Translation to Induce PD-L1 Expression for Tumor Immune Evasion. Nat. Commun. 2022, 13, 2059. [Google Scholar] [CrossRef]
- Zhao, T.; Li, Y.; Zhang, J.; Zhang, B. PD-L1 Expression Increased by IFN-γ via JAK2-STAT1 Signaling and Predicts a Poor Survival in Colorectal Cancer. Oncol. Lett. 2020, 20, 1127–1134. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Y.; Lv, X. IL4I1 Enhances PD-L1 Expression through JAK/STAT Signaling Pathway in Lung Adenocarcinoma. Immunogenetics 2022, 75, 17–25. [Google Scholar] [CrossRef]
- Black, M.; Barsoum, I.B.; Truesdell, P.; Cotechini, T.; Macdonald-Goodfellow, S.K.; Petroff, M.; Siemens, D.R.; Koti, M.; Craig, A.W.B.; Graham, C.H. Activation of the PD-1/PD-L1 Immune Checkpoint Confers Tumor Cell Chemoresistance Associated with Increased Metastasis. Oncotarget 2016, 7, 10557–10567. [Google Scholar] [CrossRef] [Green Version]
- Seetharamu, N.; Preeshagul, I.R.; Sullivan, K.M. New PD-L1 Inhibitors in Non-Small Cell Lung Cancer—Impact of Atezolizumab. Lung Cancer 2017, 8, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Guo, C.-Y.; Tou, F.-F.; Wen, X.-M.; Kuang, Y.-K.; Zhu, Q.; Hu, H. Association of PD-L1 Expression Status with the Efficacy of PD-1/PD-L1 Inhibitors and Overall Survival in Solid Tumours: A Systematic Review and Meta-Analysis. Int. J. Cancer 2020, 147, 116–127. [Google Scholar] [CrossRef]
- Liu, S.; Chen, S.; Yuan, W.; Wang, H.; Chen, K.; Li, D.; Li, D. PD-1/PD-L1 Interaction up-Regulates MDR1/P-Gp Expression in Breast Cancer Cells via PI3K/AKT and MAPK/ERK Pathways. Oncotarget 2017, 8, 99901–99912. [Google Scholar] [CrossRef] [Green Version]
- Emami, F.; Banstola, A.; Vatanara, A.; Lee, S.; Kim, J.O.; Jeong, J.-H.; Yook, S. Doxorubicin and Anti-PD-L1 Antibody Conjugated Gold Nanoparticles for Colorectal Cancer Photochemotherapy. Mol. Pharm. 2019, 16, 1184–1199. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined Cytotoxic Chemotherapy and Immunotherapy of Cancer: Modern Times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Liu, P.; Wang, K.; Glorieux, C.; Hu, Y.; Wen, S.; Jiang, W.; Huang, P. Chemotherapy Induces Tumor Immune Evasion by Upregulation of Programmed Cell Death Ligand 1 Expression in Bone Marrow Stromal Cells. Mol. Oncol. 2017, 11, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug Targeting to Tumors: Principles, Pitfalls and (Pre-) Clinical Progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Coimbra, M.; Crielaard, B.J.; Storm, G.; Schiffelers, R.M. Critical Factors in the Development of Tumor-Targeted Anti-Inflammatory Nanomedicines. J. Control. Release 2012, 160, 232–238. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Chen, G.; Zhang, J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef]
- Mishra, P.; Nayak, B.; Dey, R.K. PEGylation in Anti-Cancer Therapy: An Overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Hussain, Z.; Khan, S.; Imran, M.; Sohail, M.; Shah, S.W.A.; de Matas, M. PEGylation: A Promising Strategy to Overcome Challenges to Cancer-Targeted Nanomedicines: A Review of Challenges to Clinical Transition and Promising Resolution. Drug Deliv. Transl. Res. 2019, 9, 721–734. [Google Scholar] [CrossRef]
- Eavarone, D.A.; Yu, X.; Bellamkonda, R.V. Targeted Drug Delivery to C6 Glioma by Transferrin-Coupled Liposomes. J. Biomed. Mater. Res. 2000, 51, 10–14. [Google Scholar] [CrossRef]
- Paszko, E.; Senge, M.O. Immunoliposomes. Curr. Med. Chem. 2012, 19, 5239–5277. [Google Scholar] [CrossRef]
- Sapra, P.; Moase, E.H.; Ma, J.; Allen, T.M. Improved Therapeutic Responses in a Xenograft Model of Human B Lymphoma (Namalwa) for Liposomal Vincristine versus Liposomal Doxorubicin Targeted via Anti-CD19 IgG2a or Fab’ Fragments. Clin. Cancer Res. 2004, 10, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Sapra, P.; Allen, T.M. Internalizing Antibodies Are Necessary for Improved Therapeutic Efficacy of Antibody-Targeted Liposomal Drugs. Cancer Res. 2002, 62, 7190–7194. [Google Scholar]
- Cheng, W.W.K.; Allen, T.M. Targeted Delivery of Anti-CD19 Liposomal Doxorubicin in B-Cell Lymphoma: A Comparison of Whole Monoclonal Antibody, Fab’ Fragments and Single Chain Fv. J. Control. Release 2008, 126, 50–58. [Google Scholar] [CrossRef]
- Tedder, T.F.; Poe, J.C.; Haas, K.M. CD22: A Multifunctional Receptor That Regulates B Lymphocyte Survival and Signal Transduction. Adv. Immunol. 2005, 88, 1–50. [Google Scholar] [CrossRef]
- Tuscano, J.M.; Martin, S.M.; Ma, Y.; Zamboni, W.; O’Donnell, R.T. Efficacy, Biodistribution, and Pharmacokinetics of CD22-Targeted Pegylated Liposomal Doxorubicin in a B-Cell Non-Hodgkin’s Lymphoma Xenograft Mouse Model. Clin. Cancer Res. 2010, 16, 2760–2768. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, R.T.; Martin, S.M.; Ma, Y.; Zamboni, W.C.; Tuscano, J.M. Development and Characterization of CD22-Targeted Pegylated-Liposomal Doxorubicin (IL-PLD). Investig. New Drugs 2010, 28, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Batist, G.; Barton, J.; Chaikin, P.; Swenson, C.; Welles, L. Myocet (Liposome-Encapsulated Doxorubicin Citrate): A New Approach in Breast Cancer Therapy. Expert. Opin. Pharmacother. 2002, 3, 1739–1751. [Google Scholar] [CrossRef]
- Swenson, C.E.; Bolcsak, L.E.; Batist, G.; Guthrie, T.H.; Tkaczuk, K.H.; Boxenbaum, H.; Welles, L.; Chow, S.-C.; Bhamra, R.; Chaikin, P. Pharmacokinetics of Doxorubicin Administered i.v. as Myocet (TLC D-99; Liposome-Encapsulated Doxorubicin Citrate) Compared with Conventional Doxorubicin When given in Combination with Cyclophosphamide in Patients with Metastatic Breast Cancer. Anticancer Drugs 2003, 14, 239–246. [Google Scholar] [CrossRef]
- Mross, K.; Niemann, B.; Massing, U.; Drevs, J.; Unger, C.; Bhamra, R.; Swenson, C.E. Pharmacokinetics of Liposomal Doxorubicin (TLC-D99; Myocet) in Patients with Solid Tumors: An Open-Label, Single-Dose Study. Cancer Chemother. Pharmacol. 2004, 54, 514–524. [Google Scholar] [CrossRef]
- Dell’Olio, M.; Scalzulli, R.P.; Sanpaolo, G.; Nobile, M.; Mantuano, F.S.; La Sala, A.; D’Arena, G.; Miraglia, E.; Lucania, A.; Mastrullo, L.; et al. Non-Pegylated Liposomal Doxorubicin (Myocet®) in Patients with Poor-Risk Aggressive B-Cell Non-Hodgkin Lymphoma. Leuk. Lymphoma 2011, 52, 1222–1229. [Google Scholar] [CrossRef]
- Viale, M.; Bertone, V.; Maric, I.; Cilli, M.; Emionite, L.; Bocchini, V.; Ponzoni, M.; Fontana, V.; De Luca, F.; Rocco, M. Enhanced Therapeutic Index of Liposomal Doxorubicin Myocet Locally Delivered by Fibrin Gels in Immunodeficient Mice Bearing Human Neuroblastoma. Pharmacol. Res. 2021, 163, 105294. [Google Scholar] [CrossRef]
- Burade, V.; Bhowmick, S.; Maiti, K.; Zalawadia, R.; Ruan, H.; Thennati, R. Lipodox® (Generic Doxorubicin Hydrochloride Liposome Injection): In Vivo Efficacy and Bioequivalence versus Caelyx® (Doxorubicin Hydrochloride Liposome Injection) in Human Mammary Carcinoma (MX-1) Xenograft and Syngeneic Fibrosarcoma (WEHI 164) Mouse Models. BMC Cancer 2017, 17, 405. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Costales, A.B.; Jaffari, M.; Urbauer, D.L.; Frumovitz, M.; Kutac, C.K.; Tran, H.; Coleman, R.L. Is It Equivalent? Evaluation of the Clinical Activity of Single Agent Lipodox® Compared to Single Agent Doxil® in Ovarian Cancer Treatment. J. Oncol. Pharm. Pract. 2016, 22, 599–604. [Google Scholar] [CrossRef]
- Smith, J.A.; Mathew, L.; Burney, M.; Nyshadham, P.; Coleman, R.L. Equivalency Challenge: Evaluation of Lipodox® as the Generic Equivalent for Doxil® in a Human Ovarian Cancer Orthotropic Mouse Model. Gynecol. Oncol. 2016, 141, 357–363. [Google Scholar] [CrossRef]
- Sehouli, J.; Camara, O.; Schmidt, M.; Mahner, S.; Seipelt, G.; Otremba, B.; Schmalfeldt, B.; Tesch, H.; Lorenz-Schlüter, C.; Oskay-Ozcelik, G.; et al. Pegylated Liposomal Doxorubicin (CAELYX) in Patients with Advanced Ovarian Cancer: Results of a German Multicenter Observational Study. Cancer Chemother. Pharmacol. 2009, 64, 585–591. [Google Scholar] [CrossRef]
- Dellapasqua, S.; Trillo Aliaga, P.; Munzone, E.; Bagnardi, V.; Pagan, E.; Montagna, E.; Cancello, G.; Ghisini, R.; Sangalli, C.; Negri, M.; et al. Pegylated Liposomal Doxorubicin (Caelyx®) as Adjuvant Treatment in Early-Stage Luminal B-like Breast Cancer: A Feasibility Phase II Trial. Curr. Oncol. 2021, 28, 433. [Google Scholar] [CrossRef]
- Dou, Y.; Hynynen, K.; Allen, C. To Heat or Not to Heat: Challenges with Clinical Translation of Thermosensitive Liposomes. J. Control. Release 2017, 249, 63–73. [Google Scholar] [CrossRef]
- Besse, H.C.; Barten-van Rijbroek, A.D.; van der Wurff-Jacobs, K.M.G.; Bos, C.; Moonen, C.T.W.; Deckers, R. Tumor Drug Distribution after Local Drug Delivery by Hyperthermia, In Vivo. Cancers 2019, 11, 1512. [Google Scholar] [CrossRef] [Green Version]
- Rajappa, S.; Joshi, A.; Doval, D.C.; Batra, U.; Rajendranath, R.; Deo, A.; Biswas, G.; Bajpai, P.; Tilak, T.V.S.; Kane, S.; et al. Novel Formulations of Docetaxel, Paclitaxel and Doxorubicin in the Management of Metastatic Breast Cancer. Oncol. Lett. 2018, 16, 3757–3769. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, B.; Yue, C.; Wu, J.; Zhao, L.; Sun, D.; Wang, R. Strategies to Release Doxorubicin from Doxorubicin Delivery Vehicles. J. Drug Target. 2018, 26, 9–26. [Google Scholar] [CrossRef]
- Birngruber, T.; Raml, R.; Gladdines, W.; Gatschelhofer, C.; Gander, E.; Ghosh, A.; Kroath, T.; Gaillard, P.J.; Pieber, T.R.; Sinner, F. Enhanced Doxorubicin Delivery to the Brain Administered through Glutathione PEGylated Liposomal Doxorubicin (2B3-101) as Compared with Generic Caelyx,(®)/Doxil(®)--a Cerebral Open Flow Microperfusion Pilot Study. J. Pharm. Sci. 2014, 103, 1945–1948. [Google Scholar] [CrossRef]
- Gaillard, P.J.; Appeldoorn, C.C.M.; Dorland, R.; van Kregten, J.; Manca, F.; Vugts, D.J.; Windhorst, B.; van Dongen, G.A.M.S.; de Vries, H.E.; Maussang, D.; et al. Pharmacokinetics, Brain Delivery, and Efficacy in Brain Tumor-Bearing Mice of Glutathione Pegylated Liposomal Doxorubicin (2B3-101). PLoS ONE 2014, 9, e82331. [Google Scholar] [CrossRef] [Green Version]
- Mehrabian, A.; Vakili-Ghartavol, R.; Mashreghi, M.; Shokooh Saremi, S.; Badiee, A.; Arabi, L.; Alavizadeh, S.H.; Moosavian, S.A.; Jaafari, M.R. Preparation, Characterization, and Biodistribution of Glutathione PEGylated Nanoliposomal Doxorubicin for Brain Drug Delivery with a Post-Insertion Approach. Iran. J. Basic Med. Sci. 2022, 25, 302–312. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Lozza, I.; Torres-Suárez, A.I. Actively Targeted Nanomedicines in Breast Cancer: From Pre-Clinal Investigation to Clinic. Cancers 2022, 14, 1198. [Google Scholar] [CrossRef]
- Makwana, V.; Karanjia, J.; Haselhorst, T.; Anoopkumar-Dukie, S.; Rudrawar, S. Liposomal Doxorubicin as Targeted Delivery Platform: Current Trends in Surface Functionalization. Int. J. Pharm. 2021, 593, 120117. [Google Scholar] [CrossRef]
- Luo, R.; Li, Y.; He, M.; Zhang, H.; Yuan, H.; Johnson, M.; Palmisano, M.; Zhou, S.; Sun, D. Distinct Biodistribution of Doxorubicin and the Altered Dispositions Mediated by Different Liposomal Formulations. Int. J. Pharm. 2017, 519, 1–10. [Google Scholar] [CrossRef]
- Mastria, E.M.; Cai, L.Y.; Kan, M.J.; Li, X.; Schaal, J.L.; Fiering, S.; Gunn, M.D.; Dewhirst, M.W.; Nair, S.K.; Chilkoti, A. Nanoparticle Formulation Improves Doxorubicin Efficacy by Enhancing Host Antitumor Immunity. J. Control. Release 2018, 269, 364–373. [Google Scholar] [CrossRef]
- Hannesdóttir, L.; Tymoszuk, P.; Parajuli, N.; Wasmer, M.-H.; Philipp, S.; Daschil, N.; Datta, S.; Koller, J.-B.; Tripp, C.H.; Stoitzner, P.; et al. Lapatinib and Doxorubicin Enhance the Stat1-Dependent Antitumor Immune Response. Eur. J. Immunol. 2013, 43, 2718–2729. [Google Scholar] [CrossRef]
- Eggleton, P.; Bremer, E.; Dudek, E.; Michalak, M. Calreticulin, a Therapeutic Target? Expert Opin. Ther. Targets 2016, 20, 1137–1147. [Google Scholar] [CrossRef]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Dendritic Cells Combined with Doxorubicin Induces Immunogenic Cell Death and Exhibits Antitumor Effects for Osteosarcoma. Oncol. Lett. 2016, 11, 2169–2175. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.W.; Jiang, W.; Reich, C.F.; Pisetsky, D.S. The Extracellular Release of HMGB1 during Apoptotic Cell Death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef]
- Schiller, M.; Heyder, P.; Ziegler, S.; Niessen, A.; Claßen, L.; Lauffer, A.; Lorenz, H.-M. During Apoptosis HMGB1 Is Translocated into Apoptotic Cell-Derived Membranous Vesicles. Autoimmunity 2013, 46, 342–346. [Google Scholar] [CrossRef]
- Lee, J.-J.; Park, I.H.; Rhee, W.J.; Kim, H.S.; Shin, J.-S. HMGB1 Modulates the Balance between Senescence and Apoptosis in Response to Genotoxic Stress. FASEB J. 2019, 33, 10942–10953. [Google Scholar] [CrossRef]
- Yang, S.; Shim, M.K.; Kim, W.J.; Choi, J.; Nam, G.-H.; Kim, J.; Kim, J.; Moon, Y.; Kim, H.Y.; Park, J.; et al. Cancer-Activated Doxorubicin Prodrug Nanoparticles Induce Preferential Immune Response with Minimal Doxorubicin-Related Toxicity. Biomaterials 2021, 272, 120791. [Google Scholar] [CrossRef]
- Merino, M.; Lozano, T.; Casares, N.; Lana, H.; Troconiz, I.F.; Ten Hagen, T.L.M.; Kochan, G.; Berraondo, P.; Zalba, S.; Garrido, M.J. Dual Activity of PD-L1 Targeted Doxorubicin Immunoliposomes Promoted an Enhanced Efficacy of the Antitumor Immune Response in Melanoma Murine Model. J. Nanobiotechnol. 2021, 19, 102. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Sun, Z. Eliciting Pyroptosis to Fuel Cancer Immunotherapy: Mechanisms and Strategies. Cancer Biol. Med. 2022, 19, 948–964. [Google Scholar] [CrossRef]
- Zhu, L.; Lin, M. The Synthesis of Nano-Doxorubicin and Its Anticancer Effect. Anti-Cancer Agents Med. Chem. 2021, 21, 2466–2477. [Google Scholar] [CrossRef]
- Ibrahim, M.; Abuwatfa, W.H.; Awad, N.S.; Sabouni, R.; Husseini, G.A. Encapsulation, Release, and Cytotoxicity of Doxorubicin Loaded in Liposomes, Micelles, and Metal-Organic Frameworks: A Review. Pharmaceutics 2022, 14, 254. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. https://doi.org/10.3390/cells12040659
Kciuk M, Gielecińska A, Mujwar S, Kołat D, Kałuzińska-Kołat Ż, Celik I, Kontek R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells. 2023; 12(4):659. https://doi.org/10.3390/cells12040659
Chicago/Turabian StyleKciuk, Mateusz, Adrianna Gielecińska, Somdutt Mujwar, Damian Kołat, Żaneta Kałuzińska-Kołat, Ismail Celik, and Renata Kontek. 2023. "Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity" Cells 12, no. 4: 659. https://doi.org/10.3390/cells12040659