
Promising Perspectives of the Antiproliferative GPER Inverse Agonist ERα17p in Breast Cancer
 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. The Peptide Corresponding to the ERα Residues 295–311 Is Responsible for Apoptosis
3. Participation of GPER in the Anti-Proliferative Action of ERα17p
- ERα17p and GPER co-localize at the cytoplasmic membrane, as shown by using fluorescence microscopy, a fluorescent version of ERα17p and the anti-GPER antibody TA35133 [28];
- A GPER siRNA abrogates ERα17p’s effects [37];
- ERα17p is inactive in a GPER knockout (KO) cellular model obtained by CRISPR/Cas9 [32].
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arao, Y.; Korach, K.S. The physiological role of estrogen receptor functional domains. Essays Biochem. 2021, 65, 867–875. [Google Scholar] [CrossRef]
- Martin, M.B.; Reiter, R.; Pham, T.; Avellanet, Y.R.; Camara, J.; Lahm, M.; Pentecost, E.; Pratap, K.; Gilmore, B.A.; Divekar, S.; et al. Estrogen-like activity of metals in Mcf-7 breast cancer cells. Endocrinology 2003, 144, 2425–2436. [Google Scholar] [CrossRef] [Green Version]
- Yi, P.; Yu, X.; Wang, Z.; O’Malley, B.W. Steroid receptor-coregulator transcriptional complexes: New insights from CryoEM. Essays Biochem. 2021, 65, 857–866. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Z. Molecular cloning and purification of the protein lysine methyltransferase SMYD2 and its co-crystallization with a target peptide from estrogen receptor alpha. Methods Mol. Biol. 2022, 2418, 345–362. [Google Scholar] [PubMed]
- Habara, M.; Shimada, M. Estrogen receptor α revised: Expression, structure, function, and stability. BioEssays 2022, 44, e2200148. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.W.; Lin, V.Y.; Shupnik, M.A. Forskolin stimulates estrogen receptor (ER) α transcriptional activity and protects ER from degradation by distinct mechanisms. Int. J. Endocrinol. 2022, 2022, 7690166. [Google Scholar] [CrossRef] [PubMed]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.C.; Corbo, L. Sumoylation of the estrogen receptor α hinge region regulates its transcriptional activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef]
- Ylikomi, T.; Bocquel, M.T.; Berry, M.; Gronemeyer, H.; Chambon, P. Cooperation of proto-signals for nuclear accumulation of estrogen and progesterone receptors. EMBO J. 1992, 11, 3681–3694. [Google Scholar] [CrossRef]
- Seielstad, D.A.; Carlson, K.E.; Kushner, P.; Greene, G.L.; Katzenellenbogen, J.A. Analysis of the structural core of the human estrogen receptor ligand-binding domain by selective proteolysis/mass spectrometric analysis. Biochemistry 1995, 34, 12605–12615. [Google Scholar] [CrossRef]
- Jacquot, Y.; Gallo, D.; Leclercq, G. Estrogen receptor alpha—Identification by a modeling approach of a potential polyproline II recognizing domain within the AF-2 region of the receptor that would play a role of prime importance in its mechanism of action. J. Steroid Biochem. Mol. Biol. 2007, 104, 7690166. [Google Scholar] [CrossRef]
- Acramel, A.; Jacquot, Y. Deciphering of a putative GPER recognition domain in ERα and ERα36. Front. Endocrinol. 2022, 13, 943343. [Google Scholar] [CrossRef]
- Narwani, T.J.; Santuz, H.; Shinada, N.; Vattekatte, A.M.; Ghouzam, Y.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. Recent advances on polyproline II. Amino Acids 2017, 49, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Hoang, H.N.; Hill, T.A.; Ruiz-Gómez, G.; Diness, F.; Mason, J.M.; Wu, C.; Abbenante, G.; Shepherd, N.E.; Fairlie, D.P. Twists or turns: Stabilising alpha vs. beta turns in tetrapeptides. Chem. Sci. 2019, 10, 10595–10600. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.; Henen, M.A.; Belnou, M.; Cantrelle, F.X.; Kamah, A.; Qi, H.; Giustiniani, J.; Chambraud, B.; Baulieu, E.E.; Lippens, G.; et al. A β-turn motif in the steroid hormone receptor’s ligand-binding domains interacts with the peptidyl-prolyl isomerase (PPIase) catalytic site of the immunophilin FKBP52. Biochemistry 2016, 55, 5366–5376. [Google Scholar] [CrossRef]
- Byrne, C.; Belnou, M.; Baulieu, E.E.; Lequin, O.; Jacquot, Y. Electronic circular dichroism and nuclear magnetic resonance studies of peptides derived from the FKBP52-interacting β-turn of the hERα ligand-binding domain. Pept. Sci. 2019, 111, e24113. [Google Scholar] [CrossRef] [Green Version]
- Gallo, D.; Jacquemotte, F.; Cleeren, A.; Laïos, I.; Hadiy, S.; Rowlands, M.G.; Caille, O.; Nonclercq, D.; Laurent, G.; Jacquot, Y.; et al. Calmodulin-independent, agonistic properties of a peptide containing the calmodulin binding site of estrogen receptor α. Mol. Cell. Endocrinol. 2007, 268, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, I.; Lacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Andò, S.; Fuqua, S.A.W. Phosphorylation of the mutant K303R estrogen receptor alpha at serine 305 affects aromatase inhibitor sensitivity. Oncogene 2010, 29, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Norris, J.D.; Fan, D.; Kerner, S.A.; McDonnell, D.P. Identification of a third autonomous activation domain within the human estrogen receptor. Mol. Endocrinol. 1997, 11, 747–754. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Loele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by molecular docking of homoisoflavones from Leopoldia comosa as ligands of estrogen receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef] [Green Version]
- Gallo, D.; Haddad, I.; Laurent, G.; Vinh, J.; Jacquemotte, F.; Jacquot, Y.; Leclercq, G. Regulatory function of the P295-T311 motif of the estrogen receptor α—Does proteasomal degradation of the receptor induce emergence of peptides implicated in estrogenic responses? Nucl. Recept. Signal. 2008, 6, e007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, D.; Leclercq, G.; Haddad, J.; Vinh, J.; Castanas, E.; Kampa, M.; Pelekanou, V.; Jacquot, Y. Estrogen Receptor Alpha Polypeptide Sequence, Diagnostic and Therapeutic Applications Thereof. U.S. Patent WO 20120449229 A1, 19 April 2012. [Google Scholar]
- Gallo, D.; Jacquot, Y.; Cleeren, A.; Jacquemotte, F.; Laïos, I.; Laurent, G.; Leclercq, G. Molecular basis of agonistic activity of ERα17p, a synthetic peptide corresponding to a sequence located at the N-terminal part of the estrogen receptor α ligand binding domain. Lett. Drug Des. Discov. 2007, 4, 346–355. [Google Scholar] [CrossRef]
- Bourgoin-Voillard, S.; Fournier, F.; Afonso, C.; Jacquot, Y.; Leclercq, G.; Tabet, J.C. Calmodulin association with the synthetic ERα17p peptide investigated by mass spectrometry. Int. J. Mass Spectrom. 2011, 305, 87–94. [Google Scholar] [CrossRef]
- Carlier, L.; Byrne, C.; Miclet, E.; Bourgoin-Voillard, S.; Nicaise, M.; Tabet, J.C.; Desmadril, M.; Leclercq, G.; Lequin, O.; Jacquot, Y. Biophysical studies of the interaction between calmodulin and the R287-T311 region of human estrogen receptor α reveals an atypical binding process. Biochem. Biophys. Res. Commun. 2012, 419, 356–361. [Google Scholar] [CrossRef]
- Gallo, D.; Haddad, I.; Duvillier, H.; Jacquemotte, F.; Laios, I.; Laurent, G.; Jacquot, I.; Vinh, J.; Leclercq, G. Trophic effect in MCF-7 cells of ERα17p, a peptide corresponding to a platform regulatory motif of the estrogen receptor alpha—Underlying mechanisms. J. Steroid Biochem. Mol. Biol. 2008, 109, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.; Miclet, E.; Broutin, I.; Gallo, D.; Pelekanou, V.; Kampa, M.; Castanas, E.; Leclercq, G.; Jacquot, Y. Identification of polyproline II regions derived from the proline-rich nuclear receptor coactivators PNRC and PNRC2: New insights for ERα coactivator interactions. Chirality 2013, 25, 628–642. [Google Scholar] [CrossRef]
- Lappano, R.; Mallet, C.; Rizzuti, B.; Grande, F.; Galli, G.R.; Byrne, C.; Broutin, I.; Boudieu, L.; Eschalier, A.; Jacquot, Y.; et al. The peptide ERα17p is a GPER inverse agonist that exerts antiproliferative effects in breast cancer cells. Cells 2019, 8, 590. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.; Khemtémourian, L.; Pelekanou, V.; Kampa, M.; Leclercq, G.; Sagan, S.; Castanas, E.; Burlina, F.; Jacquot, Y. ERα17p, a peptide reproducing the hinge region of the estrogen receptor α associates to biological membranes: A biophysical approach. Steroids 2012, 77, 979–987. [Google Scholar] [CrossRef]
- Trichet, M.; Lappano, R.; Belnou, M.; Vazquez, L.S.S.; Alves, I.; Ravault, D.; Sagan, S.; Khemtemourian, L.; Maggiolini, M.; Jacquot, Y. Interaction of the anti-proliferative GPER inverse agonist ERα17p with the breast cancer cell plasma membrane: From biophysics to biology. Cells 2019, 9, 447. [Google Scholar] [CrossRef] [Green Version]
- Miclet, E.; Bourgoin-Voillard, S.; Byrne, C.; Jacquot, Y. Application of circular dichroism spectroscopy to the analysis of the interaction between the estrogen receptor alpha and coactivators: The case of calmodulin. Methods Mol. Biol. 2016, 1366, 241–259. [Google Scholar]
- Jouffre, B.; Acramel, A.; Belnou, M.; Santolla, M.F.; Talia, M.; Lappano, R.; Nemati, F.; Decaudin, D.; Khemtemourian, L.; Liu, W.Q.; et al. Identification of a human estrogen receptor α tetrapeptidic fragment with dual antiproliferative and anti-nociceptive action. Sci. Rep. 2023, 13, 1326. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, F.S.; Byrne, C.; Khemtemourian, L.; Ducouret, G.; Dietler, G.; Jacquot, Y. Concentration-dependent and surface-assisted self-assembly properties of a bioactive estrogen receptor α-derived peptide. J. Pept. Sci. 2015, 21, 95–104. [Google Scholar] [CrossRef]
- Yip, F.; Nemati, F.; El Botty, R.; Belnou, M.; Decaudin, D.; Mansuy, C.; Jacquot, Y. Improvement of the anti-proliferative activity of the peptide ERα17p in MCF-7 breast cancer cells using nanodiamonds. Ann. Pharm. Fr. 2019, 77, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Pelekanou, V.M.; Kampa, D.; Gallo, G.; Notas, M.; Troullinaki, H.; Duvillier, Y.; Jacquot, E.N.; Stathopoulos, E. Castanas and G. Leclercq. The estrogen receptor α-derived peptide ERα17p (P(295)-T(311)) exerts pro-apoptotic actions in breast cancer cells in vitro and in vivo, independently from their ERα status. Mol. Oncol. 2011, 5, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notas, G.; Kampa, M.; Pelekanou, V.; Troullinaki, M.; Jacquot, Y.; Leclercq, G.; Castanas, E. Whole transcriptome analysis of the ERα synthetic fragment P295-T311 (ERα17p) identifies specific ERα-isoform (ERα, ERα36)-dependent and -independent actions in breast cancer cells. Mol. Oncol. 2013, 7, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Leiber, D.; Burlina, F.; Byrne, C.; Robin, P.; Piesse, C.; Gonzalez, L.; Leclercq, G.; Tanfin, Z.; Jacquot, Y. The sequence Pro295-Thr311 of the hinge region of oestrogen receptor α is involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem. J. 2015, 472, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Kampa, M.V.; Pelekanou, D.; Gallo, G.; Notas, M.; Troullinaki, I.; Pediaditakis, I.; Charalampopoulos, Y.; Jacquot, G. Leclercq and E. Castanas. ERα17p, an ERα P295-T311 fragment, modifies the migration of breast cancer cells, through actin cytoskeleton rearrangements. J. Cell. Biochem. 2011, 112, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Kalyvianaki, K.; Panagiotopoulos, A.A.; Patentalaki, M.; Castanas, E.; Kampa, M. Importins involved in the nuclear transportation of steroid hormone receptors: In silico and in vitro data. Front. Endocrinol. 2022, 13, 954629. [Google Scholar] [CrossRef]
- Kaynak, A.; Davis, H.W.; Kogan, A.B.; Lee, J.H.; Narmoneva, D.A.; Qi, X. Phosphatidylserine: The unique dual-role biomarker for cancer imaging and therapy. Cancers 2022, 14, 2536. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.; Quinn, J.; Pang, Y.; Graeber, C.; Shaw, S.; Dong, J.; Thomas, P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007, 148, 3236–3245. [Google Scholar] [CrossRef] [Green Version]
- Pupo, M.; Vivacqua, A.; Perrotta, I.; Pisano, A.; Aquila, S.; Abonante, S.; Gasperi-Campani, A.; Pezzi, V.; Maggiolini, M. The nuclear localization signal is required for nuclear GPER translocation and function in breast Cancer-Associated Fibroblasts (CAFs). Mol. Cell. Endocrinol. 2013, 25, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.H.; Chu, N.M.; Lin, Y.F.; Kao, S.H. G-protein coupled estrogen receptor in breast cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Liu, D. Does GPER really function as a G protein-coupled estrogen receptor in vivo? Front. Endocrinol. 2020, 11, 148. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER mediates a feedforward FGF2/FGFR1 paracrine activation coupling CAFs to cancer cells toward breast tumor progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Marjon, N.A.; Hu, C.; Hathaway, H.J.; Prossnitz, E.R. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol. Cancer Res. 2014, 12, 1644–1654. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef] [Green Version]
- Arias-Pulido, H.; Royce, M.; Gong, Y.; Joste, N.; Lomo, L.; Lee, S.J.; Chaher, N.; Verschraegen, C.; Lara, J.; Prossnitz, E.R.; et al. GPR30 and estrogen receptor expression: New insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res. Treat. 2010, 123, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Talia, M.; De Francesco, E.M.; Rigiracciolo, D.C.; Muoio, M.G.; Muglia, L.; Belfiore, A.; Maggiolini, M.; Sims, A.H.; Lappano, R. The G Protein-coupled estrogen receptor (GPER) expression correlates with pro-metastatic pathways in ER-negative breast cancer: A Bioinformatics Analysis. Cells 2020, 9, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappano, R.; Maggiolini, M. GPER is involved in the functional liaison between breast tumor cells and cancer-associated fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 2018, 176, 49–56. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic perspectives on the modulation of G-protein coupled estrogen receptor, GPER, function. Front. Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef]
- Lappano, R.; Jacquot, Y.; Maggiolini, M. GPCR modulation in breast cancer. Int. J. Mol. Sci. 2018, 19, 3840. [Google Scholar] [CrossRef] [Green Version]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Arakaki, A.K.S.; Pan, W.A.; Trejo, J. GPCRs in cancer: Protease-activated receptors, endocytic adaptors and signaling. Int. J. Mol. Sci. 2018, 19, 1886. [Google Scholar] [CrossRef]
- Kondakova, I.V.; Shashova, E.E.; Sidenko, E.A.; Astakhova, T.M.; Zakharova, L.A.; Sharova, N.P. Estrogen receptors and ubiquitin proteasome system: Mutual regulation. Biomolecules 2020, 10, 500. [Google Scholar] [CrossRef] [Green Version]
- Mallet, C.; Boudieu, L.; Lamoine, S.; Coudert, C.; Jacquot, Y.; Eschalier, A. ERα17p exerts anti-hyperlagesic and anti-inflammatory actions through GPER in mice. Front. Endocrinol. 2021, 12, 794332. [Google Scholar] [CrossRef]
- Maggiolini, M.; Santolla, M.F.; Avino, S.; Aiello, F.; Rosano, C.; Garofalo, A.; Grande, F. Identification of two benzopyrroloxazines acting as selective GPER antagonists in breast cancer cells and cancer-associated fibroblasts. Future Med. Chem. 2015, 7, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, V.; Ramirez-Sanchez, I.; Moreno-Ulloa, A.; Romero-Perez, D.; Chávez, D.; Ortiz, M.; Najera, N.; Correa-Basurto, J.; Villarreal, F.; Ceballos, G. Synthesis of novel (–)-epicatechin derivatives as potential endothelial GPER agonists: Evaluation of biological effects. Bioorganic Med. Chem. Lett. 2018, 28, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Muñoz, A.; Prestegui-Martel, B.; Méndez-Luna, D.; Fragoso-Vázquez, M.J.; García-Sánchez, J.R.; Bello, M.; Martínez-Archundia, M.; Chávez-Blanco, A.; Dueñas-González, A.; Mendoza-Lujambio, I.; et al. Selection of a GPER1 ligand via ligand-based virtual screening coupled to molecular dynamics simulations and its anti-proliferative effects on breast cancer cells. Anticancer Agents Med. Chem. 2018, 18, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Kezimana, P.; Dmitriev, A.A.; Kudryavtseva, A.V.; Romanova, E.V.; Melnikova, N.V. Secoisolariciresinol diglucoside of flaxseed and its metabolites: Biosynthesis and potential for nutraceuticals. Front. Genet. 2018, 9, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, F.; Lappano, R.; Bruno, L.; Rizzuti, B.; Grande, F.; Guzzi, R.; Briguori, S.; Miglietta, A.M.; Nakajima, M.; Di Martino, M.T.; et al. AHR and GPER mediate the stimulatory effects induced by 3-methylcholanthrene in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2019, 38, 335. [Google Scholar] [CrossRef] [Green Version]
- Bello, M.; Méndez-Luna, D.; Sarmiento, V.; Correa Basurto, J.; Najera, N.; Villarreal, F.; Ceballos, G. Structural and energetic basis for novel epicatechin derivatives acting as GPER agonists through the MMGBSA method. J. Steroid Biochem. Mol. Biol. 2019, 189, 176–186. [Google Scholar] [CrossRef]
- Zacarías-Lara, O.J.; Méndez-Luna, D.; Martínez-Ruíz, G.; García-Sanchéz, J.R.; Fragoso-Vázquez, M.J.; Bello, M.; Becerra-Martínez, E.; García-Vázquez, J.B.; Correa-Basurto, J. Synthesis and in vitro evaluation of tetrahydroquinoline derivatives as antiproliferative compounds of breast cancer via targeting the GPER. Anticancer Agents Med. Chem. 2019, 19, 760–771. [Google Scholar] [CrossRef]
- Grande, F.; Occhiuzzi, M.A.; Lappano, R.; Cirillo, F.; Guzzi, R.; Garofalo, A.; Jacquot, Y.; Maggiolini, M.; Rizzuti, B. Computational approaches for the discovery of GPER targeting compounds. Front. Endocrinol. 2020, 11, 517. [Google Scholar] [CrossRef]



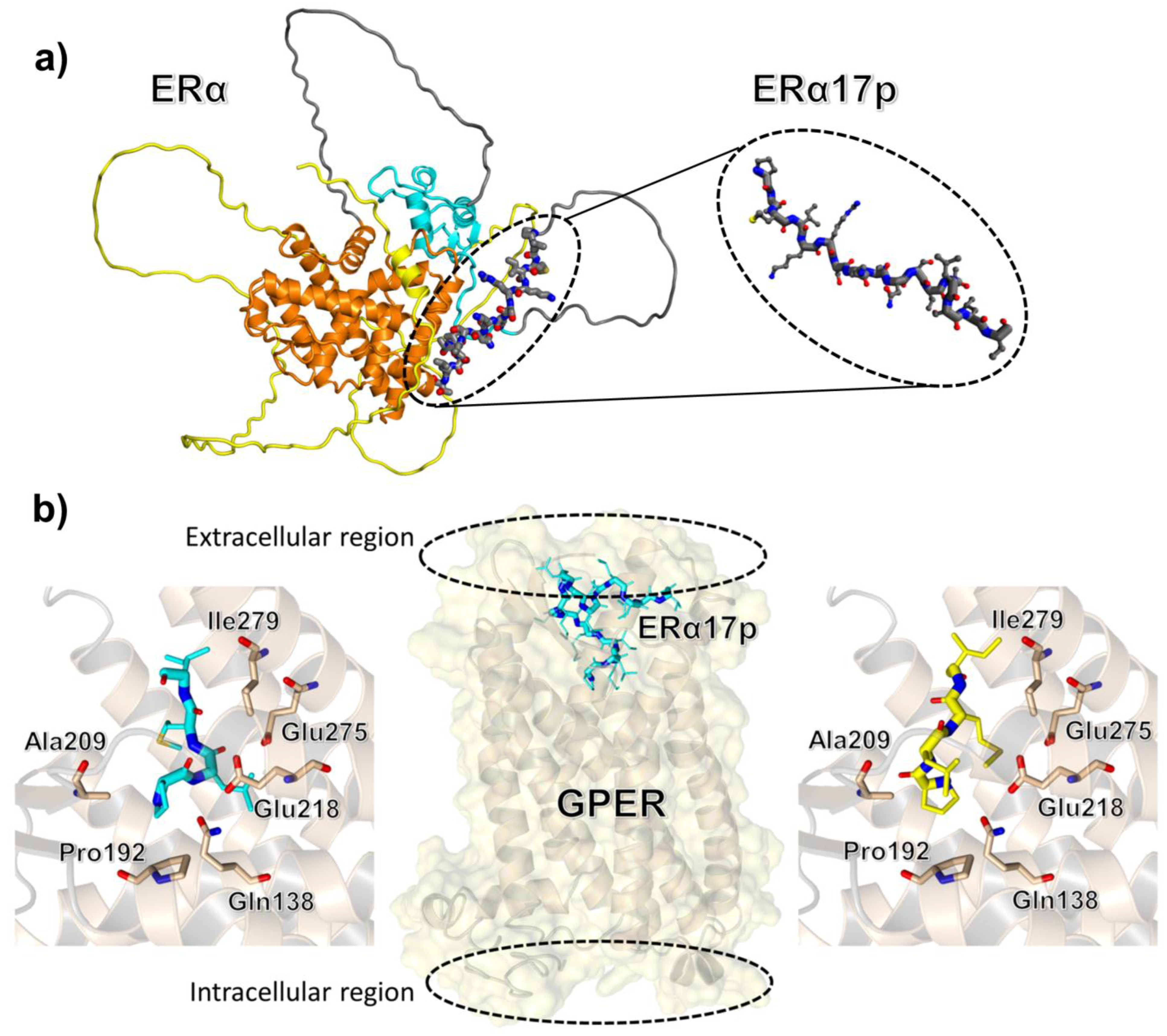{kind=link}
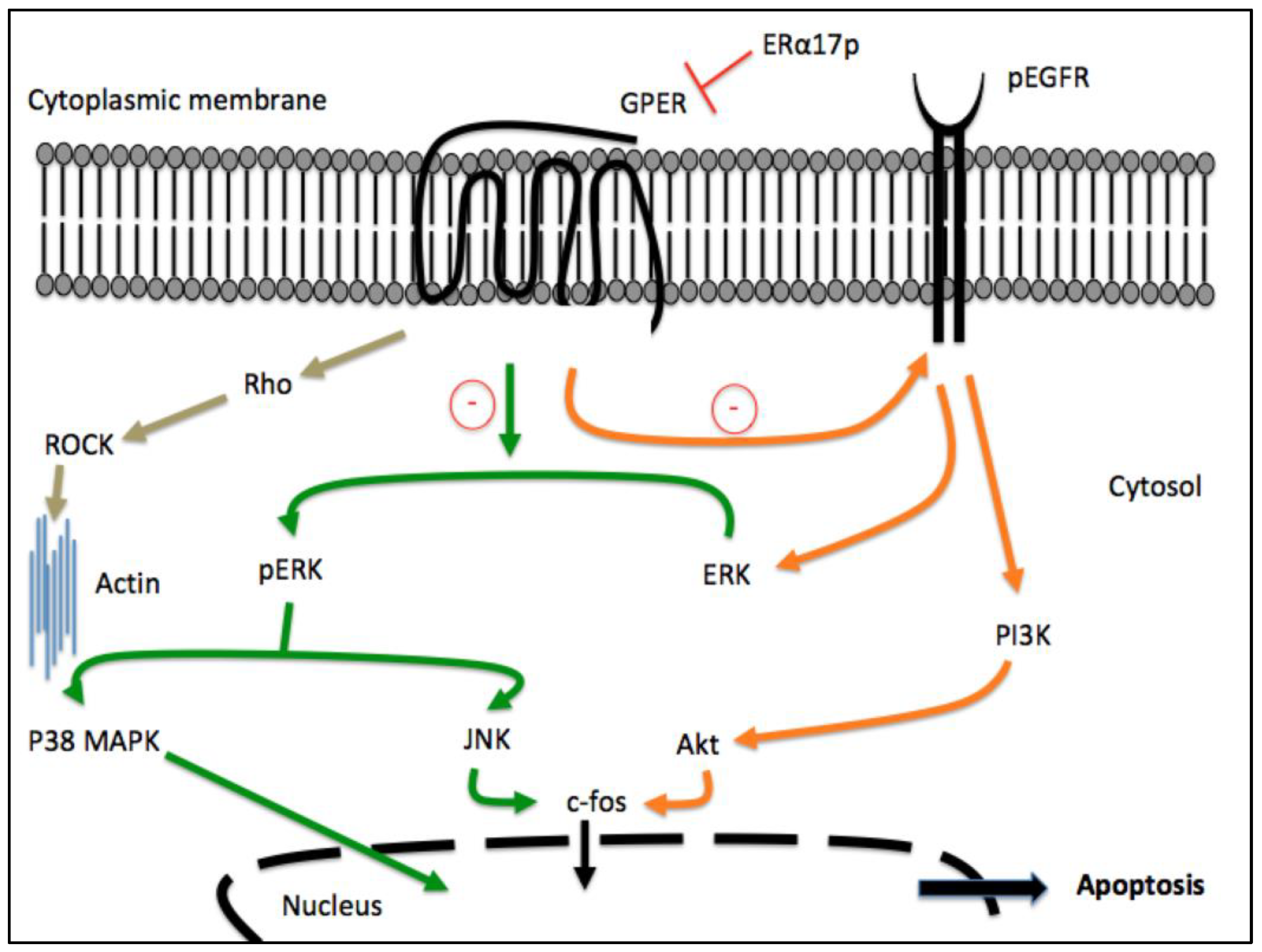{kind=link}
| Interaction partners of the 295–311 region of ERα (in the context of the whole protein) |
| Ca2+-calmodulin [16] |
| Direct partners of the 295–311 region of ERα (in the context of the peptide ERα17p) |
| Ca2+-calmodulin [16,24,25,31] ERα17p, to form amyloid fibrils, hydrogels and complex aggregates [30,32,33] Estrogen receptor α [21] GPER [28] Heat Shock Protein 70 (HSP70) [26] Hard and soft negative lipid-containing surfaces including cell membrane models [29,33,34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kampa, M.; Lappano, R.; Grande, F.; Rizzuti, B.; Maggiolini, M.; Castanas, E.; Jacquot, Y. Promising Perspectives of the Antiproliferative GPER Inverse Agonist ERα17p in Breast Cancer. Cells 2023, 12, 653. https://doi.org/10.3390/cells12040653
Kampa M, Lappano R, Grande F, Rizzuti B, Maggiolini M, Castanas E, Jacquot Y. Promising Perspectives of the Antiproliferative GPER Inverse Agonist ERα17p in Breast Cancer. Cells. 2023; 12(4):653. https://doi.org/10.3390/cells12040653
Chicago/Turabian StyleKampa, Marilena, Rosamaria Lappano, Fedora Grande, Bruno Rizzuti, Marcello Maggiolini, Elias Castanas, and Yves Jacquot. 2023. "Promising Perspectives of the Antiproliferative GPER Inverse Agonist ERα17p in Breast Cancer" Cells 12, no. 4: 653. https://doi.org/10.3390/cells12040653