
Emerging Mechanisms of Skeletal Muscle Homeostasis and Cachexia: The SUMO Perspective


{kind=link}
{kind=link}
Abstract
:1. Introduction
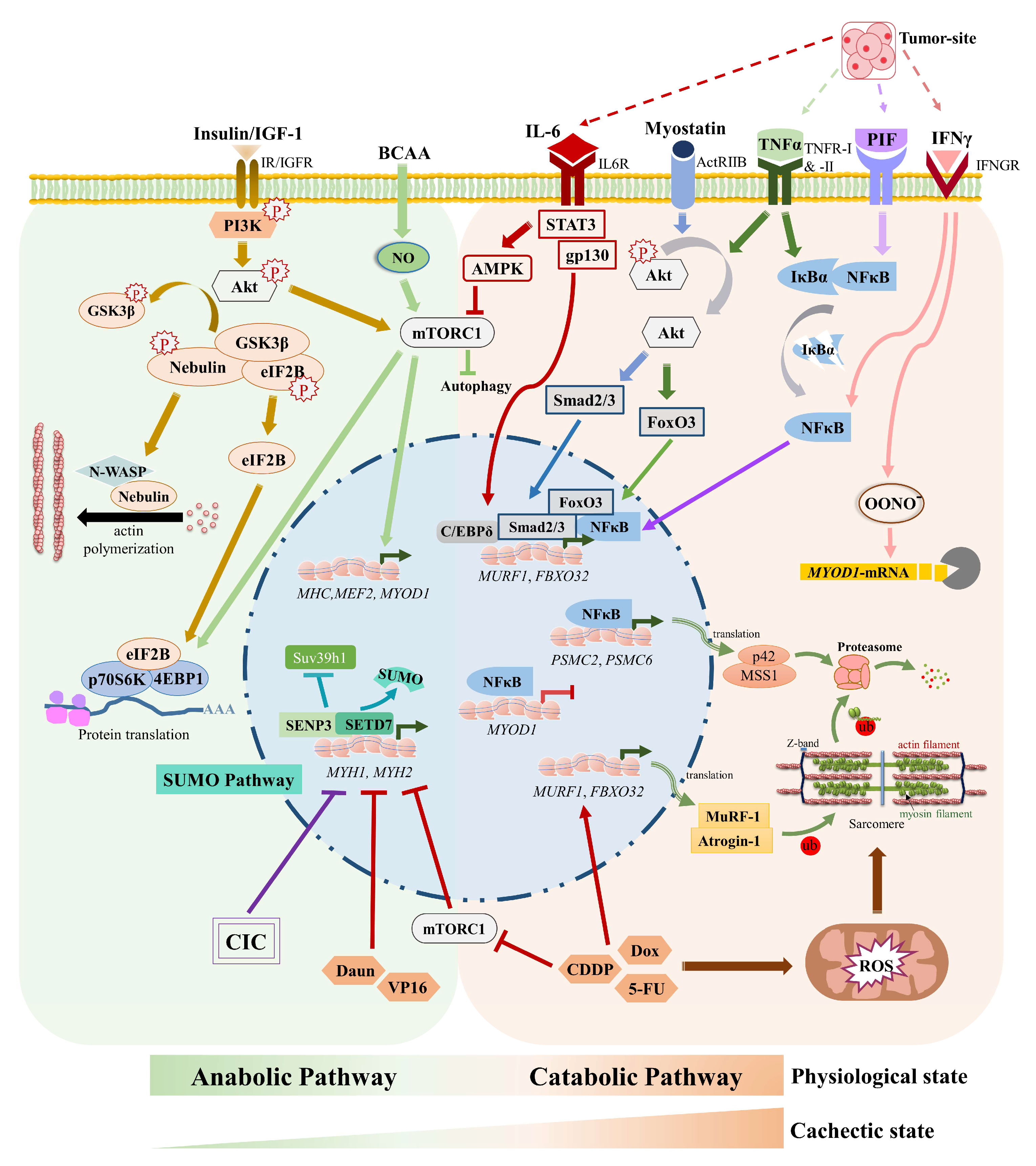
2. Cachexia Affects Muscle Cell Signaling Mechanisms and Metabolism
2.1. Cachexia and Muscle-Specific E3 Ubiquitin Ligases
2.2. Cachexia and Akt-mTOR Signaling
2.3. Cachexia, Glucose Uptake & Metabolism
2.4. Cachexia and the Vicious Cori Cycle
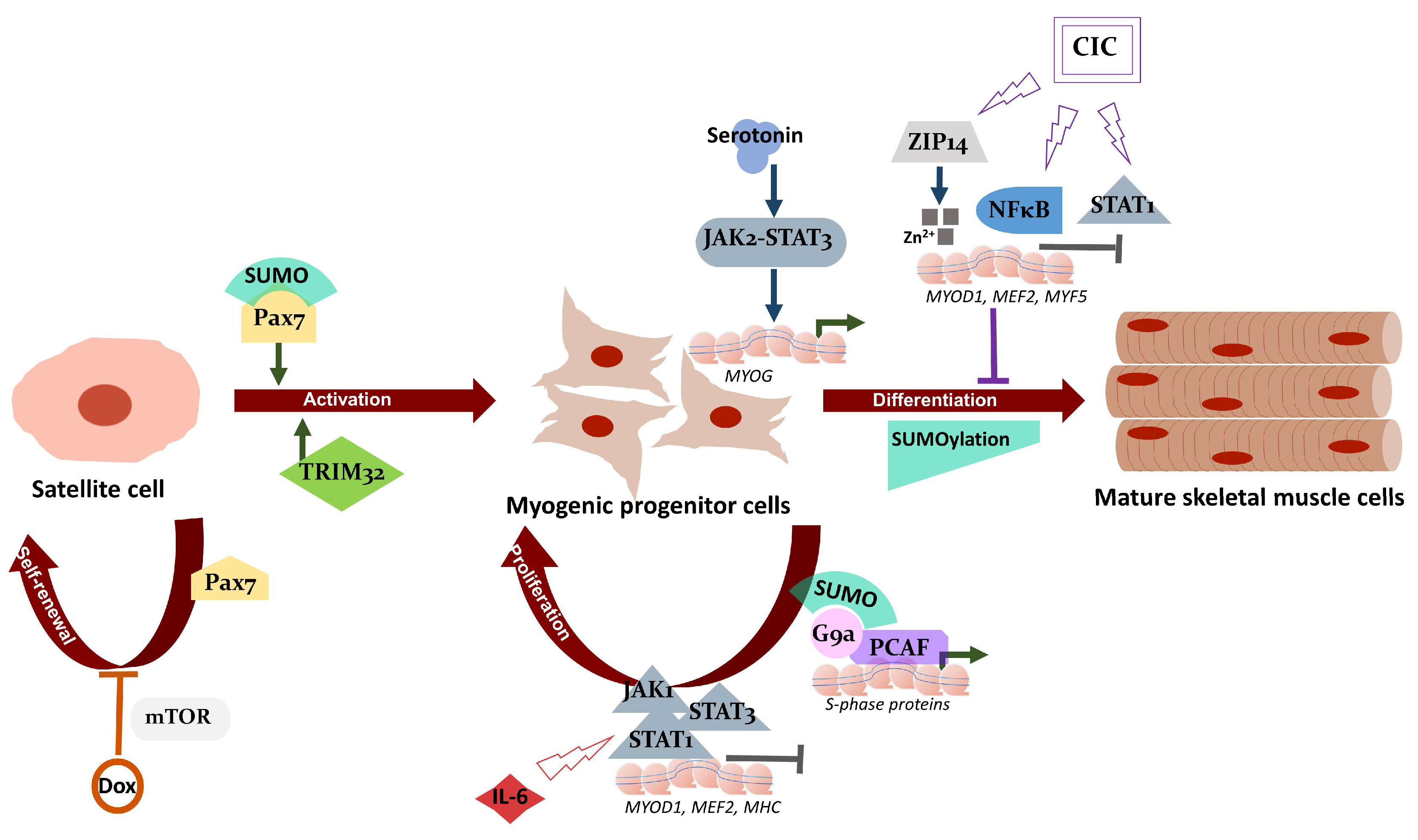
2.5. JAK/STAT Pathway and Cachexia
2.6. Cachexia, Zinc Homeostasis, and Satellite Cell Differentiation
3. SUMO Pathway, Muscle Physiology, and Cachexia
3.1. The SUMO Pathway
3.2. SUMOs in Muscle Physiology
3.3. SUMOs in Muscle Atrophy including Cachexia
4. Current Treatment Strategies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley, A.F.; Niedergerke, R. Structural changes in muscle during contraction; interference microscopy of living muscle fibres. Nature 1954, 173, 971–973. [Google Scholar] [CrossRef]
- Moore, D.R. Keeping older muscle “young” through dietary protein and physical activity. Adv. Nutr. 2014, 5, 599S–607S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Gunnarsson, R.; Bjorkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Investig. 1985, 76, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, V.; Bernetti, A.; Mangone, M.; Paoloni, M. Clinical definition of sarcopenia. Clin. Cases Miner. Bone Metab. 2014, 11, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J. Reversing sarcopenia: How weight training can build strength and vitality. Geriatrics 1996, 51, 46–47. [Google Scholar] [PubMed]
- Ali, S.; Garcia, J.M. Sarcopenia, cachexia and aging: Diagnosis, mechanisms and therapeutic options—A mini-review. Gerontology 2014, 60, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Gould, D.W.; Lahart, I.; Carmichael, A.R.; Koutedakis, Y.; Metsios, G.S. Cancer cachexia prevention via physical exercise: Molecular mechanisms. J. Cachexia Sarcopenia Muscle 2013, 4, 111–124. [Google Scholar] [CrossRef]
- Murphy, B.T.; Mackrill, J.J.; O’Halloran, K.D. Impact of cancer cachexia on respiratory muscle function and the therapeutic potential of exercise. J. Physiol. 2022, 600, 4979–5004. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 2019, 10, 140–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Yoneda, J.; Ohmori, H.; Sasaki, T.; Shimbo, K.; Eto, S.; Kato, Y.; Miyano, H.; Kobayashi, T.; Sasahira, T.; et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 2014, 74, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Damrauer, J.S.; Stadler, M.E.; Acharyya, S.; Baldwin, A.S.; Couch, M.E.; Guttridge, D.C. Chemotherapy-induced muscle wasting: Association with NF-kappaB and cancer cachexia. Eur. J. Transl. Myol. 2018, 28, 7590. [Google Scholar] [CrossRef] [Green Version]
- Katz, A.M.; Katz, P.B. Diseases of the heart in the works of Hippocrates. Br. Heart J. 1962, 24, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Bennani-Baiti, N.; Walsh, D. What is cancer anorexia-cachexia syndrome? A historical perspective. J. R. Coll Physicians Edinb. 2009, 39, 257–262. [Google Scholar]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Amrute-Nayak, M.; Pegoli, G.; Holler, T.; Lopez-Davila, A.J.; Lanzuolo, C.; Nayak, A. Chemotherapy triggers cachexia by deregulating synergetic function of histone-modifying enzymes. J. Cachexia Sarcopenia Muscle 2020, 12, 159–176. [Google Scholar] [CrossRef]
- von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. Cachexia Sarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef] [PubMed]
- Malinin, N.L.; Boldin, M.P.; Kovalenko, A.V.; Wallach, D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature 1997, 385, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Investig. 2004, 114, 370–378. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wang, X.; Gao, T.; Tian, H.; Zhou, D.; Zhang, L.; Li, G.; Wang, X. The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia. Am. J. Clin. Nutr. 2020, 111, 570–579. [Google Scholar] [CrossRef]
- Segatto, M.; Fittipaldi, R.; Pin, F.; Sartori, R.; Dae Ko, K.; Zare, H.; Fenizia, C.; Zanchettin, G.; Pierobon, E.S.; Hatakeyama, S.; et al. Epigenetic targeting of bromodomain protein BRD4 counteracts cancer cachexia and prolongs survival. Nat. Commun. 2017, 8, 1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyke, S.M.; Tisdale, M.J. NF-kappaB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin-proteasome system in skeletal muscle. Br. J. Cancer 2005, 92, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khal, J.; Wyke, S.M.; Russell, S.T.; Hine, A.V.; Tisdale, M.J. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br. J. Cancer 2005, 93, 774–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attaix, D.; Taillandier, D.; Combaret, L.; Ralliere, C.; Larbaud, D.; Aurousseau, E.; Tanaka, K. Expression of subunits of the 19S complex and of the PA28 activator in rat skeletal muscle. Mol. Biol. Rep. 1997, 24, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Lagirand, J.; Batonnet, S.; Sirri, V.; Leibovitch, M.P.; Leibovitch, S.A. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 2005, 280, 2847–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.; Schauer, A.; Augstein, A.; Kirchhoff, V.; Draskowski, R.; Jannasch, A.; Goto, K.; Lyall, G.; Mannel, A.; Barthel, P.; et al. Targeting MuRF1 by small molecules in a HFpEF rat model improves myocardial diastolic function and skeletal muscle contractility. J. Cachexia Sarcopenia Muscle 2022, 13, 1565–1581. [Google Scholar] [CrossRef]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef]
- Adams, V.; Gussen, V.; Zozulya, S.; Cruz, A.; Moriscot, A.; Linke, A.; Labeit, S. Small-Molecule Chemical Knockdown of MuRF1 in Melanoma Bearing Mice Attenuates Tumor Cachexia Associated Myopathy. Cells 2020, 9, 2272. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, G.; Zhang, Z.; Zhu, J.Z.; Li, L.; Zhou, Y.; Rodney, G.G., Jr.; Abo-Zahrah, R.S.; Anderson, L.; Garcia, J.M.; et al. UBR2 targets myosin heavy chain IIb and IIx for degradation: Molecular mechanism essential for cancer-induced muscle wasting. Proc. Natl. Acad. Sci. USA 2022, 119, e2200215119. [Google Scholar] [CrossRef]
- Dworzak, F.; Ferrari, P.; Gavazzi, C.; Maiorana, C.; Bozzetti, F. Effects of cachexia due to cancer on whole body and skeletal muscle protein turnover. Cancer 1998, 82, 42–48. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Attenuation of muscle atrophy in a murine model of cachexia by inhibition of the dsRNA-dependent protein kinase. Br. J. Cancer 2007, 96, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet Muscle 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geremia, A.; Sartori, R.; Baraldo, M.; Nogara, L.; Balmaceda, V.; Dumitras, G.A.; Ciciliot, S.; Scalabrin, M.; Nolte, H.; Blaauw, B. Activation of Akt-mTORC1 signalling reverts cancer-dependent muscle wasting. J. Cachexia Sarcopenia Muscle 2022, 13, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.A.; McNally, R.M.; Hoffmann, F.M.; Hornberger, T.A. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 2013, 27, 1946–1957. [Google Scholar] [CrossRef]
- Herningtyas, E.H.; Okimura, Y.; Handayaningsih, A.E.; Yamamoto, D.; Maki, T.; Iida, K.; Takahashi, Y.; Kaji, H.; Chihara, K. Branched-chain amino acids and arginine suppress MaFbx/atrogin-1 mRNA expression via mTOR pathway in C2C12 cell line. Biochim. Biophys. Acta 2008, 1780, 1115–1120. [Google Scholar] [CrossRef]
- Kobayashi, H.; Borsheim, E.; Anthony, T.G.; Traber, D.L.; Badalamenti, J.; Kimball, S.R.; Jefferson, L.S.; Wolfe, R.R. Reduced amino acid availability inhibits muscle protein synthesis and decreases activity of initiation factor eIF2B. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E488–E498. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, K.; Sun, L.; Jiao, H.; Zhou, Y.; Li, H.; Wang, X.; Zhao, J.; Lin, H. L-Arginine/nitric oxide regulates skeletal muscle development via muscle fibre-specific nitric oxide/mTOR pathway in chickens. Anim. Nutr. 2022, 10, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; Gao, S.; Narsale, A.A.; Carson, J.A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 2014, 28, 998–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Honors, M.A.; Kinzig, K.P. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J. Cachexia Sarcopenia Muscle 2012, 3, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 2010, 126, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Parekh, N.; Lin, Y.; Hayes, R.B.; Albu, J.B.; Lu-Yao, G.L. Longitudinal associations of blood markers of insulin and glucose metabolism and cancer mortality in the third National Health and Nutrition Examination Survey. Cancer Causes Control 2010, 21, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.L.; Tian, M.; Kliewer, K.L.; Belury, M.A. Rosiglitazone delayed weight loss and anorexia while attenuating adipose depletion in mice with cancer cachexia. Cancer Biol. Ther. 2011, 12, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [Green Version]
- Mannelli, M.; Gamberi, T.; Magherini, F.; Fiaschi, T. A Metabolic Change towards Fermentation Drives Cancer Cachexia in Myotubes. Biomedicines 2021, 9, 698. [Google Scholar] [CrossRef]
- Rubin, R.P. Carl and Gerty Cori: A collaboration that changed the face of biochemistry. J. Med. Biogr. 2021, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, R.J.; Kammerer, U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 2011, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J. Effects of 2-deoxyglucose on carbohydrate metablism: Review of the literature and studies in the rat. Metabolism 1962, 11, 1098–1112. [Google Scholar]
- Zhu, Z.; Jiang, W.; McGinley, J.N.; Thompson, H.J. 2-Deoxyglucose as an energy restriction mimetic agent: Effects on mammary carcinogenesis and on mammary tumor cell growth in vitro. Cancer Res. 2005, 65, 7023–7030. [Google Scholar] [CrossRef] [Green Version]
- Guillet-Deniau, I.; Burnol, A.F.; Girard, J. Identification and localization of a skeletal muscle secrotonin 5-HT2A receptor coupled to the Jak/STAT pathway. J. Biol. Chem. 1997, 272, 14825–14829. [Google Scholar] [CrossRef] [Green Version]
- Moresi, V.; Adamo, S.; Berghella, L. The JAK/STAT Pathway in Skeletal Muscle Pathophysiology. Front. Physiol. 2019, 10, 500. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ma, K.; Wang, H.; Xiao, F.; Gao, Y.; Zhang, W.; Wang, K.; Gao, X.; Ip, N.; Wu, Z. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 2007, 179, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Wang, H.; Fu, X.; Li, Y.; Ma, K.; Sun, L.; Gao, X.; Wu, Z. Oncostatin M inhibits myoblast differentiation and regulates muscle regeneration. Cell Res. 2011, 21, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wang, C.; Xiao, F.; Wang, H.; Wu, Z. JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 2008, 283, 34029–34036. [Google Scholar] [CrossRef] [Green Version]
- Silva, K.A.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.M.; Wu, C.L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 2014, 14, 997. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Zhong, L.; Zhu, J.; Xu, H.; Ma, W.; Zhang, L.; Shen, Y.; Law, B.Y.; Ding, F.; Gu, X.; et al. Inhibition of IL-6/JAK/STAT3 pathway rescues denervation-induced skeletal muscle atrophy. Ann. Transl. Med. 2020, 8, 1681. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Liu, Z.; Bharadwaj, U.; Wang, H.; Wang, X.; Zhang, S.; Liuzzi, J.P.; Chang, S.M.; Cousins, R.J.; et al. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc. Natl. Acad. Sci. USA 2007, 104, 18636–18641. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice with Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; Lomicronpez-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Shakri, A.R.; James Zhong, T.; Ma, W.; Coker, C.; Hegde, R.; Scholze, H.; Chin, V.; Szabolcs, M.; Hibshoosh, H.; Tanji, K.; et al. Aberrant Zip14 expression in muscle is associated with cachexia in a Bard1-deficient mouse model of breast cancer metastasis. Cancer Med. 2020, 9, 6766–6775. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, S.; Mazroui, R.; Dallaire, P.; Chittur, S.; Tenenbaum, S.A.; Radzioch, D.; Marette, A.; Gallouzi, I.E. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol. Cell Biol. 2005, 25, 6533–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Guttridge, D.C. Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin. Cancer Res. 2007, 13, 1356–1361. [Google Scholar] [CrossRef] [Green Version]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, R.; Gerace, L.; Melchior, F. Molecular characterization of the SUMO-1 modification of RanGAP1 and its role in nuclear envelope association. J. Cell Biol. 1998, 140, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Müller, S. SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol. 2014, 15, 422. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wansleeben, C.; Zhao, S.; Miao, P.; Paschen, W.; Yang, W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014, 15, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Vertegaal, A.C.O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell Biol. 2022, 23, 715–731. [Google Scholar] [CrossRef]
- de The, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2021, 22, 215–235. [Google Scholar] [CrossRef]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Fatouros, C.; Lee, H.; Eisenhardt, N. SUMO conjugation—A mechanistic view. Biomol. Concepts 2017, 8, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, F.; de Robertis, O.; Laviola, L.; Montrone, C.; Perrini, S.; McCowen, K.C.; Smith, R.J. The sentrin-conjugating enzyme mUbc9 interacts with GLUT4 and GLUT1 glucose transporters and regulates transporter levels in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1125–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalioti, V.S.; Vergarajauregui, S.; Pulido, D.; Sandoval, I.V. The insulin-sensitive glucose transporter, GLUT4, interacts physically with Daxx. Two proteins with capacity to bind Ubc9 and conjugated to SUMO1. J. Biol. Chem. 2002, 277, 19783–19791. [Google Scholar] [CrossRef] [Green Version]
- Ariga, M.; Nedachi, T.; Katagiri, H.; Kanzaki, M. Functional role of sortilin in myogenesis and development of insulin-responsive glucose transport system in C2C12 myocytes. J. Biol. Chem. 2008, 283, 10208–10220. [Google Scholar] [CrossRef] [Green Version]
- Koo, Y.D.; Choi, J.W.; Kim, M.; Chae, S.; Ahn, B.Y.; Kim, M.; Oh, B.C.; Hwang, D.; Seol, J.H.; Kim, Y.B.; et al. SUMO-Specific Protease 2 (SENP2) Is an Important Regulator of Fatty Acid Metabolism in Skeletal Muscle. Diabetes 2015, 64, 2420–2431. [Google Scholar] [CrossRef] [Green Version]
- Koo, Y.D.; Lee, J.S.; Lee, S.A.; Quaresma, P.G.F.; Bhat, R.; Haynes, W.G.; Park, Y.J.; Kim, Y.B.; Chung, S.S.; Park, K.S. SUMO-specific protease 2 mediates leptin-induced fatty acid oxidation in skeletal muscle. Metabolism 2019, 95, 27–35. [Google Scholar] [CrossRef]
- Riquelme, C.; Barthel, K.K.; Liu, X. SUMO-1 modification of MEF2A regulates its transcriptional activity. J. Cell Mol. Med. 2006, 10, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Gregoire, S.; Yang, X.J. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol. Cell Biol. 2005, 25, 2273–2287. [Google Scholar] [CrossRef] [Green Version]
- Hietakangas, V.; Anckar, J.; Blomster, H.A.; Fujimoto, M.; Palvimo, J.J.; Nakai, A.; Sistonen, L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 2006, 103, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riquelme, C.; Barthel, K.K.; Qin, X.F.; Liu, X. Ubc9 expression is essential for myotube formation in C2C12. Exp. Cell Res. 2006, 312, 2132–2141. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lee, S.M.; Joung, H. 2-D08 treatment regulates C2C12 myoblast proliferation and differentiation via the Erk1/2 and proteasome signaling pathways. J. Muscle Res. Cell Motil. 2021, 42, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.C.; Fedorov, Y.V.; Rosenthal, R.S.; Olwin, B.B. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J. Cell Physiol. 2001, 186, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Oustanina, S.; Hause, G.; Braun, T. Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. 2004, 23, 3430–3439. [Google Scholar] [CrossRef] [Green Version]
- Luan, Z.; Liu, Y.; Stuhlmiller, T.J.; Marquez, J.; Garcia-Castro, M.I. SUMOylation of Pax7 is essential for neural crest and muscle development. Cell Mol. Life Sci. 2013, 70, 1793–1806. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Shankar, S.R.; Wang, Y.; Taneja, R. SUMOylation of G9a regulates its function as an activator of myoblast proliferation. Cell Death Dis. 2019, 10, 250. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Namuduri, A.V.; Heras, G.; Mi, J.; Cacciani, N.; Hornaeus, K.; Konzer, A.; Lind, S.B.; Larsson, L.; Gastaldello, S. A Proteomic Approach to Identify Alterations in the Small Ubiquitin-like Modifier (SUMO) Network during Controlled Mechanical Ventilation in Rat Diaphragm Muscle. Mol. Cell. Proteomics 2017, 16, 1081–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namuduri, A.V.; Heras, G.; Lauschke, V.M.; Vitadello, M.; Traini, L.; Cacciani, N.; Gorza, L.; Gastaldello, S. Expression of SUMO enzymes is fiber type dependent in skeletal muscles and is dysregulated in muscle disuse. FASEB J. 2020, 34, 2269–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadena, S.M.; Zhang, Y.; Fang, J.; Brachat, S.; Kuss, P.; Giorgetti, E.; Stodieck, L.S.; Kneissel, M.; Glass, D.J. Skeletal muscle in MuRF1 null mice is not spared in low-gravity conditions, indicating atrophy proceeds by unique mechanisms in space. Sci. Rep. 2019, 9, 9397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baehr, L.M.; Tunzi, M.; Bodine, S.C. Muscle hypertrophy is associated with increases in proteasome activity that is independent of MuRF1 and MAFbx expression. Front. Physiol. 2014, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [Green Version]
- Nayak, A.; Lopez-Davila, A.J.; Kefalakes, E.; Holler, T.; Kraft, T.; Amrute-Nayak, M. Regulation of SETD7 Methyltransferase by SENP3 Is Crucial for Sarcomere Organization and Cachexia. Cell Rep. 2019, 27, 2725–2736.e4. [Google Scholar] [CrossRef] [Green Version]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef]
- Campelj, D.G.; Goodman, C.A.; Rybalka, E. Chemotherapy-Induced Myopathy: The Dark Side of the Cachexia Sphere. Cancers 2021, 13, 3615. [Google Scholar] [CrossRef]
- Braun, T.P.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.J.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Moylan, J.S.; Patterson, E.W.; Smith, J.D.; Wilson, A.S.; Rabbani, Z.; Reid, M.B. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2012, 302, C195–C202. [Google Scholar] [CrossRef] [Green Version]
- D’Lugos, A.C.; Fry, C.S.; Ormsby, J.C.; Sweeney, K.R.; Brightwell, C.R.; Hale, T.M.; Gonzales, R.J.; Angadi, S.S.; Carroll, C.C.; Dickinson, J.M. Chronic doxorubicin administration impacts satellite cell and capillary abundance in a muscle-specific manner. Physiol. Rep. 2019, 7, e14052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulmi, J.J.; Nissinen, T.A.; Rasanen, M.; Degerman, J.; Lautaoja, J.H.; Hemanthakumar, K.A.; Backman, J.T.; Ritvos, O.; Silvennoinen, M.; Kivela, R. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Hain, B.A.; Xu, H.; Waning, D.L. Loss of REDD1 prevents chemotherapy-induced muscle atrophy and weakness in mice. J. Cachexia Sarcopenia Muscle 2021, 12, 1597–1612. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Barber, M.D.; Moses, A.G.; Ahmedzai, S.H.; Taylor, G.S.; Tisdale, M.J.; Murray, G.D. Double-blind, placebo-controlled, randomized study of eicosapentaenoic acid diester in patients with cancer cachexia. J. Clin. Oncol. 2006, 24, 3401–3407. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Calder, P.C.; Schols, A.; Lonnqvist, F.; Bech, M.; Muscaritoli, M. Safety and Tolerability of Targeted Medical Nutrition for Cachexia in Non-Small-Cell Lung Cancer: A Randomized, Double-Blind, Controlled Pilot Trial. Nutr. Cancer 2020, 72, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.J.; Mukerji, P.; Tisdale, M.J. Attenuation of proteasome-induced proteolysis in skeletal muscle by [148]-hydroxy-[148]-methylbutyrate in cancer-induced muscle loss. Cancer Res. 2005, 65, 277–283. [Google Scholar] [CrossRef] [PubMed]
- May, P.E.; Barber, A.; D’Olimpio, J.T.; Hourihane, A.; Abumrad, N.N. Reversal of cancer-related wasting using oral supplementation with a combination of beta-hydroxy-beta-methylbutyrate, arginine, and glutamine. Am. J. Surg. 2002, 183, 471–479. [Google Scholar] [CrossRef]
- Prado, C.M.; Orsso, C.E.; Pereira, S.L.; Atherton, P.J.; Deutz, N.E.P. Effects of beta-hydroxy beta-methylbutyrate (HMB) supplementation on muscle mass, function, and other outcomes in patients with cancer: A systematic review. J. Cachexia Sarcopenia Muscle 2022, 13, 1623–1641. [Google Scholar] [CrossRef]
- Thambamroong, T.; Seetalarom, K.; Saichaemchan, S.; Pumsutas, Y.; Prasongsook, N. Efficacy of Curcumin on Treating Cancer Anorexia-Cachexia Syndrome in Locally or Advanced Head and Neck Cancer: A Double-Blind, Placebo-Controlled Randomised Phase IIa Trial (CurChexia). J. Nutr. Metab. 2022, 2022, 5425619. [Google Scholar] [CrossRef]
- Kapoor, N.; Naufahu, J.; Tewfik, S.; Bhatnagar, S.; Garg, R.; Tewfik, I. A Prospective Randomized Controlled Trial to Study the Impact of a Nutrition-Sensitive Intervention on Adult Women With Cancer Cachexia Undergoing Palliative Care in India. Integr. Cancer Ther. 2017, 16, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.M.; Boccia, R.V.; Graham, C.D.; Yan, Y.; Duus, E.M.; Allen, S.; Friend, J. Anamorelin for patients with cancer cachexia: An integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials. Lancet Oncol. 2015, 16, 108–116. [Google Scholar] [CrossRef]
- Katakami, N.; Uchino, J.; Yokoyama, T.; Naito, T.; Kondo, M.; Yamada, K.; Kitajima, H.; Yoshimori, K.; Sato, K.; Saito, H.; et al. Anamorelin (ONO-7643) for the treatment of patients with non-small cell lung cancer and cachexia: Results from a randomized, double-blind, placebo-controlled, multicenter study of Japanese patients (ONO-7643-04). Cancer 2018, 124, 606–616. [Google Scholar] [CrossRef]
- Currow, D.; Temel, J.S.; Abernethy, A.; Milanowski, J.; Friend, J.; Fearon, K.C. ROMANA 3: A phase 3 safety extension study of anamorelin in advanced non-small-cell lung cancer (NSCLC) patients with cachexia. Ann. Oncol. 2017, 28, 1949–1956. [Google Scholar] [CrossRef]
- Queiroz, A.L.; Dantas, E.; Ramsamooj, S.; Murthy, A.; Ahmed, M.; Zunica, E.R.M.; Liang, R.J.; Murphy, J.; Holman, C.D.; Bare, C.J.; et al. Blocking ActRIIB and restoring appetite reverses cachexia and improves survival in mice with lung cancer. Nat. Commun. 2022, 13, 4633. [Google Scholar] [CrossRef]
- Ranjbar, K.; Ballaro, R.; Bover, Q.; Pin, F.; Beltra, M.; Penna, F.; Costelli, P. Combined Exercise Training Positively Affects Muscle Wasting in Tumor-Bearing Mice. Med. Sci. Sports Exerc. 2019, 51, 1387–1395. [Google Scholar] [CrossRef]
- Ballaro, R.; Penna, F.; Pin, F.; Gomez-Cabrera, M.C.; Vina, J.; Costelli, P. Moderate Exercise Improves Experimental Cancer Cachexia by Modulating the Redox Homeostasis. Cancers 2019, 11, 285. [Google Scholar] [CrossRef] [Green Version]
- Padilha, C.S.; Borges, F.H.; Costa Mendes da Silva, L.E.; Frajacomo, F.T.T.; Jordao, A.A.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; Deminice, R. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro-inflammatory state, and cachexia in Walker-256 tumor-bearing rats. Appl. Physiol. Nutr. Metab. 2017, 42, 916–923. [Google Scholar] [CrossRef]
- Tanaka, M.; Sugimoto, K.; Fujimoto, T.; Xie, K.; Takahashi, T.; Akasaka, H.; Kurinami, H.; Yasunobe, Y.; Matsumoto, T.; Fujino, H.; et al. Preventive effects of low-intensity exercise on cancer cachexia-induced muscle atrophy. FASEB J. 2019, 33, 7852–7862. [Google Scholar] [CrossRef]
- Gatta, L.; Vitiello, L.; Gorini, S.; Chiandotto, S.; Costelli, P.; Giammarioli, A.M.; Malorni, W.; Rosano, G.; Ferraro, E. Modulating the metabolism by trimetazidine enhances myoblast differentiation and promotes myogenesis in cachectic tumor-bearing c26 mice. Oncotarget 2017, 8, 113938–113956. [Google Scholar] [CrossRef] [Green Version]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musaro, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Sanchez, A.M.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, A.J.; Silva, V.; Sawaris Neto, L.; Teixeira Basmage, J.P.; Peccin, M.S.; Maddocks, M. Exercise for cancer cachexia in adults. Cochrane Database Syst. Rev. 2021, 3, CD010804. [Google Scholar] [CrossRef]
- Bourdel-Marchasson, I.; Blanc-Bisson, C.; Doussau, A.; Germain, C.; Blanc, J.F.; Dauba, J.; Lahmar, C.; Terrebonne, E.; Lecaille, C.; Ceccaldi, J.; et al. Nutritional advice in older patients at risk of malnutrition during treatment for chemotherapy: A two-year randomized controlled trial. PLoS ONE 2014, 9, e108687. [Google Scholar] [CrossRef]
- Clemente-Suarez, V.J.; Redondo-Florez, L.; Rubio-Zarapuz, A.; Martinez-Guardado, I.; Navarro-Jimenez, E.; Tornero-Aguilera, J.F. Nutritional and Exercise Interventions in Cancer-Related Cachexia: An Extensive Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 4604. [Google Scholar] [CrossRef]
- Shahriyari, M.; Islam, M.R.; Sakib, S.M.; Rinn, M.; Rika, A.; Kruger, D.; Kaurani, L.; Gisa, V.; Winterhoff, M.; Anandakumar, H.; et al. Engineered skeletal muscle recapitulates human muscle development, regeneration and dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 3106–3121. [Google Scholar] [CrossRef]
- Nayak, A.; Amrute-Nayak, M. SUMO system—A key regulator in sarcomere organization. FEBS J. 2020, 287, 2176–2190. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, B.; Gand, L.V.; Amrute-Nayak, M.; Nayak, A. Emerging Mechanisms of Skeletal Muscle Homeostasis and Cachexia: The SUMO Perspective. Cells 2023, 12, 644. https://doi.org/10.3390/cells12040644
Khan B, Gand LV, Amrute-Nayak M, Nayak A. Emerging Mechanisms of Skeletal Muscle Homeostasis and Cachexia: The SUMO Perspective. Cells. 2023; 12(4):644. https://doi.org/10.3390/cells12040644
Chicago/Turabian StyleKhan, Bushra, Luis Vincens Gand, Mamta Amrute-Nayak, and Arnab Nayak. 2023. "Emerging Mechanisms of Skeletal Muscle Homeostasis and Cachexia: The SUMO Perspective" Cells 12, no. 4: 644. https://doi.org/10.3390/cells12040644