
Molecular Drivers of Myelodysplastic Neoplasms (MDS)—Classification and Prognostic Relevance


Abstract
:1. Introduction
2. Classification of MDS
2.1. French–American–British (FAB) Classification
2.2. WHO Classification
- MDS with defining genetic abnormalities;
- ○
- MDS with low blasts and isolated 5q deletion (MDS-5q);
- ○
- MDS with low blasts and SF3B1 mutation (MDS-SF3B1);
- ○
- MDS with biallelic TP53 inactivation (MDS-biTP53);
- MDS, morphologically defined;
- ○
- MDS with low blasts (MDS-LB);
- ○
- MDS, hypoplastic (MDS-h);
- ○
- MDS with increased blasts (MDS-IB);
- ▪
- MDS-IB1;
- ▪
- MDS-IB2;
- ▪
- MDS with fibrosis (MDS-f).
2.3. International Consensus Classification (ICC)
- MDS with mutated SF3B1 (MDS-SF3B1);
- MDS with del(5q) [MDS-del5q)];
- MDS, NOS without dysplasia;
- MDS, NOS with single lineage dysplasia;
- MDS, NOS with multilineage dysplasia;
- MDS with excess blasts (MDS-EB);
- MDS/AML.
2.4. Internal Prognostic Scoring System (IPSS)
3. Cytogenetic and Molecular Landscape of MDS
3.1. Recurrent Cytogenetic Abnormalities in MDS
3.1.1. Deletion 5q
3.1.2. Monosomy 7 and Deletion 7q
3.1.3. Trisomy 8
3.1.4. Deletion 20q
3.1.5. Other Cytogenetic Abnormalities
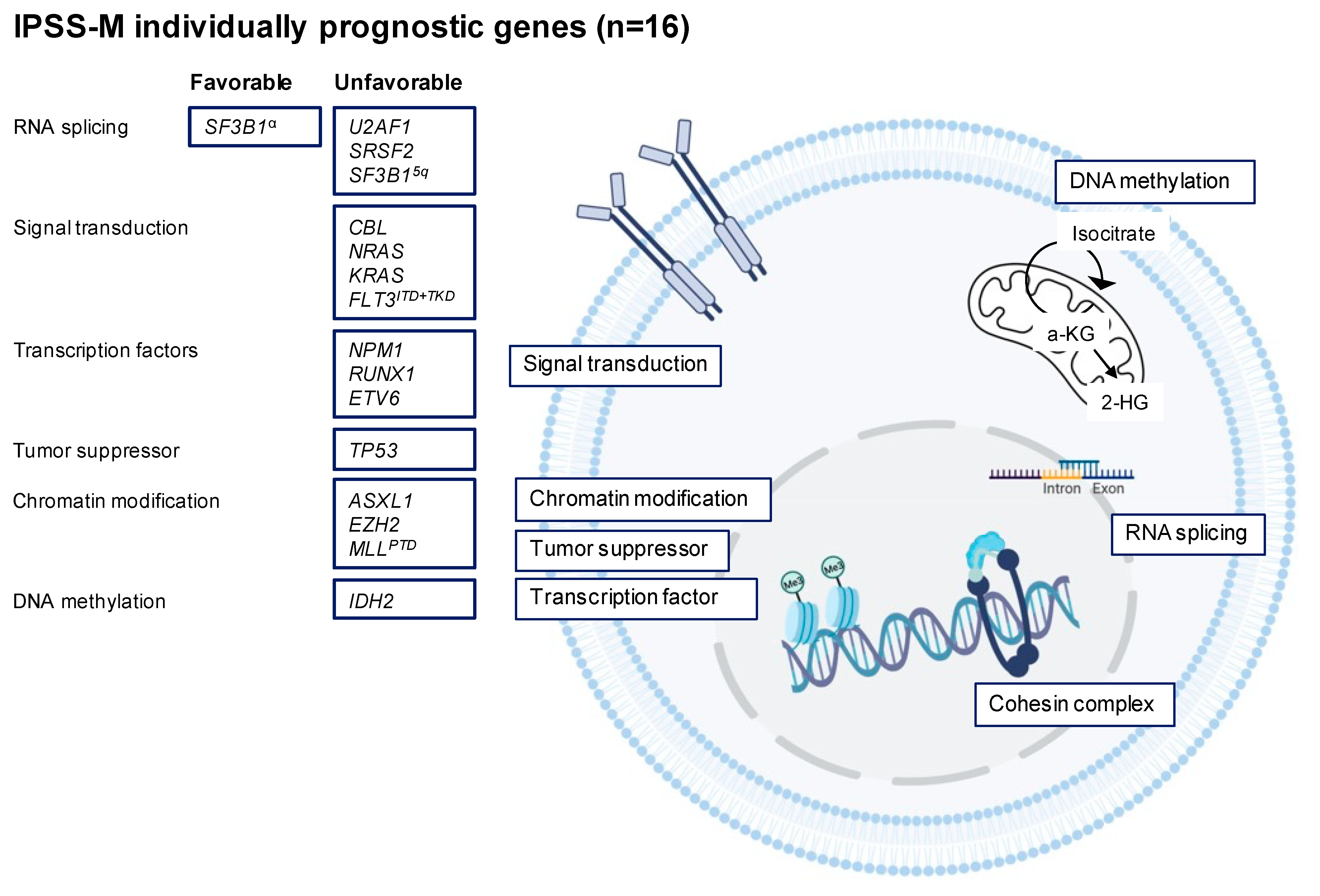
3.2. Recurrent Gene Mutations in MDS
3.2.1. RNA Splicing
3.2.2. DNA Methylation
3.2.3. Chromatin Modification
3.2.4. Transcription Factors
3.2.5. Cohesin Complex
3.2.6. Signal Transduction
3.2.7. TP53
4. Conclusions
5. Future Directions

{kind=link}
| Frequency (%) | Location | Prognostic Impact | Function | Ref | |
|---|---|---|---|---|---|
| RNA splicing (40–50%) | |||||
| SF3B1 | 25–30% Frequently associated with MDS-RS | 2q33 | Favorable Unfavorable if combined with del 5q. | Subunit 1, RNA-splicing factor 3b complex, part of U2 small nuclear ribonucleoprotein complex (snRNP) | [10,40,41] |
| SRSF2 | 15% Higher frequently in chronic myelomonocytic leukemia (50%) | 17q25 | Unfavorable OS, high-risk transformation into AML | Serine/arginine (SR) rich splicing factor 2, family of pre-mRNA splicing factors | [10,44,75] |
| U2AF1 | 10–15% | 21q22 | Unfavorable OS, high-risk transformation into AML | Heteromeric with U2AF2 to form U2 auxiliary factor (U2AF), recruits U2 snRNP. Pre-mRNA splicing factor. | [10,46] |
| ZRSR2 | 5–10% | Xp22 | Unclear | Zinc finger RNA-binding associated with U2. 3′ intron splice site recognition. | [44,76] |
| U2AF2 | Rare | 19q13 | Unfavorable, associated high-risk MDS and AML | Heteromeric with U2AF1 that forms U2AF | [77,78] |
| DNA methylation (30–40%) | |||||
| TET2 | 20–30% | 4q24 | Unclear | Alpha ketoglutarate-dependent dioxygenase | [10,15,17] |
| DNMT3A | 15% | 2q23 | Unfavorable, associated risk transformation into AML | DNA methyltransferase 3A, catalyzes transfer methyl groups to cytosine residue in CpG dinucleotides | [10,17] |
| IDH2 | 5% | 2q33 | Unclear, studies suggest unfavorable prognosis | NADPH-dependent isocitrate dehydrogenase | [10,69] |
| IDH1 | 2% | 15q26 | Unfavorable | NADPH-dependent isocitrate dehydrogenase | [79,80] |
| Chromatin modification (20%) | |||||
| ASXL1 | 15–20% | 20q11 | Unfavorable | Polycomb group protein, chromatin-binding protein | [10,38,56] |
| EZH2 | 5–10% | 7q36 | Unfavorable | Polycomb group protein, histone methyl transferase | [15,38,59] |
| KDM6A | <5% | Xp11 | Unclear | Polycomb group protein, lysine demethylation | [10] |
| EED | <5% | 11q14 | Unclear | Polycomb group protein, histone methyl transferase | [10] |
| Transcription factors (10–15%) | |||||
| RUNX1 | 10% | 21q22 | Unfavorable | Transcription factor, core-binding factor complex | [10,15,16] |
| BCOR | 5% | Xp11 | Unfavorable | Transcription factor, polycomb complex protein | [10,15,16,63] |
| ETV6 | <5% | 12p13 | Unfavorable | ETS family transcription factor | [10,15] |
| GATA2 | <5% | 3q21 | Unfavorable | Zinc finger transcription factor | [10,62] |
| Cohesin (10%) | |||||
| STAG2 | 5% | Xq25 | Unfavorable | Component cohesin complex | [10,68] |
| RAD21 | <5% | 8q24 | Unclear | Component cohesin complex | [68] |
| Signal transduction (5–10%) | |||||
| JAK2 | <5% | 9p24 | Unclear | Tyrosine kinase, JAK-STAT pathway | [15] |
| CBL | 5% | 11q23 | Unfavorable | Tyrosine kinase, E3 ubiquitin-protein ligase | [15,70] |
| NRAS | <5% | 1q13 | Unfavorable | Tyrosine kinase, RAS-MAPK pathway | [10,81] |
| KRAS | <5% | 12p12 | Unfavorable | Tyrosine kinase, RAS-MAPK pathway | [10,81] |
| FLT3-ITD | <5% | 13q12 | Unfavorable | Class III family receptor tyrosine kinase | [10] |
| KIT | <5% | 4q11-12 | Unclear | Class III family receptor tyrosine kinase | [10] |
| PTPN11 | <5% | 12q24 | Unclear | Protein phosphatase | [16] |
| Tumor suppressor (5–10%) | |||||
| TP53 | 10%, 50% in complex karyotype | 17p13 | Unfavorable | Tumor suppressor, transcription factor | [10,74] |
| WT1 | 5% | 11p13 | Unfavorable, associated with disease progression to AML | Tumor suppressor, transcription factor | [10,82] |
| PHF6 | 5% | Xq26-27 | Unfavorable | Tumor suppressor, epigenetic transcriptional regulator | [83] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.N.; Bejar, R. MDS overlap disorders and diagnostic boundaries. Blood 2019, 133, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol. 1982, 51, 189–199. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J.; Lister, T.A.; Bloomfield, C.D. World Health Organization Classification of Neoplastic Diseases of the Hematopoietic and Lymphoid Tissues: Report of the Clinical Advisory Committee Meeting—Airlie House, Virginia, November 1997. J. Clin. Oncol. 1999, 17, 3835–3849. [Google Scholar] [CrossRef] [Green Version]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Malcovati, L.; Galli’, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, P.; Cox, C.; Lebeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International Scoring System for Evaluating Prognosis in Myelodysplastic Syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Benton, C.B.; Khan, M.; Sallman, D.; Nazha, A.; Nogueras González, G.M.; Piao, J.; Ning, J.; Aung, F.; Al Ali, N.; Jabbour, E.; et al. Prognosis of patients with intermediate risk IPSS-R myelodysplastic syndrome indicates variable outcomes and need for models beyond IPSS-R. Am. J. Hematol. 2018, 93, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeilstöcker, M.; Tuechler, H.; Sanz, G.; Schanz, J.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Time-dependent changes in mortality and transformation risk in MDS. Blood 2016, 128, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [Green Version]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Levine, R.; Ebert, B.L. Unraveling the Molecular Pathophysiology of Myelodysplastic Syndromes. J. Clin. Oncol. 2011, 29, 504–515. [Google Scholar] [CrossRef]
- Sperling, A.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New Comprehensive Cytogenetic Scoring System for Primary Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia After MDS Derived From an International Database Merge. J. Clin. Oncol. 2012, 30, 820–829. [Google Scholar] [CrossRef]
- List, A.; Ebert, B.L.; Fenaux, P. A decade of progress in myelodysplastic syndrome with chromosome 5q deletion. Leukemia 2018, 32, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, H.; Cassiman, J.-J.; David, G.; Fryns, J.-P.; Michaux, J.-L.; Sokal, G. Distinct haematological disorder with deletion of long arm of No. 5 chromosome. Nature 1974, 251, 437–438. [Google Scholar] [CrossRef] [PubMed]
- Gurnari, C.; Piciocchi, A.; Soddu, S.; Bonanni, F.; Scalzulli, E.; Niscola, P.; Di Veroli, A.; Piccioni, A.L.; Piedimonte, M.; Maiorana, G.; et al. Myelodysplastic syndromes with del(5q): A real-life study of determinants of long-term outcomes and response to lenalidomide. Blood Cancer J. 2022, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Zeidan, A.M. Transforming growth factor (TGF)-β pathway as a therapeutic target in lower risk myelodysplastic syndromes. Leukemia 2019, 33, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.K.; Ademà, V.; Heckl, D.; Järås, M.; Mallo, M.; Lord, A.M.; Chu, L.P.; McConkey, M.E.; Kramann, R.; Mullally, A.; et al. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of del(5q) MDS. Cancer Cell 2014, 26, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Inaba, T.; Honda, H.; Matsui, H. The enigma of monosomy 7. Blood 2018, 131, 2891–2898. [Google Scholar] [CrossRef]
- Dolnik, A.; Engelmann, J.C.; Scharfenberger-Schmeer, M.; Mauch, J.; Kelkenberg-Schade, S.; Haldemann, B.; Fries, T.; Krönke, J.; Kühn, M.W.M.; Paschka, P.; et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 2012, 120, e83–e92. [Google Scholar] [CrossRef] [Green Version]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; Van Der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; De Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Starý, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Drevon, L.; Marceau, A.; Maarek, O.; Cuccuini, W.; Clappier, E.; Eclache, V.; Cluzeau, T.; Richez, V.; Berkaoui, I.; Dimicoli-Salazar, S.; et al. Myelodysplastic syndrome (MDS) with isolated trisomy 8: A type of MDS frequently associated with myeloproliferative features? A report by the Groupe Francophone des Myélodysplasies. Br. J. Haematol. 2018, 182, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloand, E.M.; Pfannes, L.; Chen, G.; Shah, S.; Solomou, E.E.; Barrett, J.; Young, N.S. CD34 cells from patients with trisomy 8 myelodysplastic syndrome (MDS) express early apoptotic markers but avoid programmed cell death by up-regulation of antiapoptotic proteins. Blood 2007, 109, 2399–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-C.; Ito, Y.; Hsiao, H.-H.; Sashida, G.; Kodama, A.; Ohyashiki, J.H.; Ohyashiki, K. Risk factor analysis in myelodysplastic syndrome patients with del(20q): Prognosis revisited. Cancer Genet. Cytogenet. 2006, 171, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Villamón, E.; Abellán, R.; Calasanz, M.J.; Irigoyen, A.; Sanz, G.; Such, E.; Mora, E.; Gutiérrez, M.; Collado, R.; et al. Myelodysplastic syndromes with 20q deletion: Incidence, prognostic value and impact on response to azacitidine of ASXL1 chromosomal deletion and genetic mutations. Br. J. Haematol. 2021, 194, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Solé, F.; Espinet, B.; Sanz, G.F.; Cervera, J.; Calasanz, M.J.; Luño, E.; Prieto, F.; Granada, I.; Hernández, J.M.; Cigudosa, J.C.; et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Br. J. Haematol. 2000, 108, 346–356. [Google Scholar] [CrossRef]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Grau, J.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. Monosomal karyotype in MDS: Explaining the poor prognosis? Leukemia 2013, 27, 1988–1995. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Maciejewski, J.P. The functional mechanisms of mutations in myelodysplastic syndrome. Leukemia 2019, 33, 2779–2794. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Huber, S.; Haferlach, T.; Meggendorfer, M.; Hutter, S.; Hoermann, G.; Baer, C.; Kern, W.; Haferlach, C. SF3B1 mutated MDS: Blast count, genetic co-abnormalities and their impact on classification and prognosis. Leukemia 2022, 36, 2894–2902. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Mortera-Blanco, T.; Dimitriou, M.; Woll, P.S.; Karimi, M.; Elvarsdottir, E.; Conte, S.; Tobiasson, M.; Jansson, M.; Douagi, I.; Moarii, M.; et al. SF3B1-initiating mutations in MDS-RSs target lymphomyeloid hematopoietic stem cells. Blood 2017, 130, 881–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kade, S.; Schlarmann, C.; Löffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.; Kölking, B.; Wichmann, M.; Görlich, K.; et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Cai, W.; Hua, Y.; Yang, X.; Zhou, J. The Biological and Clinical Consequences of RNA Splicing Factor U2AF1 Mutation in Myeloid Malignancies. Cancers 2022, 14, 4406. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Midic, D.; Rinke, J.; Perner, F.; Müller, V.; Hinze, A.; Pester, F.; Landschulze, J.; Ernst, J.; Gruhn, B.; Matziolis, G.; et al. Prevalence and dynamics of clonal hematopoiesis caused by leukemia-associated mutations in elderly individuals without hematologic disorders. Leukemia 2020, 34, 2198–2205. [Google Scholar] [CrossRef]
- Jawad, M.; Afkhami, M.; Ding, Y.; Zhang, X.; Li, P.; Young, K.; Xu, M.L.; Cui, W.; Zhao, Y.; Halene, S.; et al. DNMT3A R882 Mutations Confer Unique Clinicopathologic Features in MDS Including a High Risk of AML Transformation. Front. Oncol. 2022, 12, 561. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Foran, J.M.; Watts, J.M.; Stein, E.M.; de Botton, S.; Fathi, A.T.; Prince, G.T.; Stein, A.S.; Stone, R.M.; Patel, P.A.; et al. MDS-265: Ivosidenib (IVO) in Patients with IDH1-Mutant Relapsed/Refractory Myelodysplastic Syndrome (R/R MDS): Updated Enrollment of a Phase 1 Dose Escalation and Expansion Study. J. Clin. Oncol. 2022, 20, S321. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Venugopal, S.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Montalban-Bravo, G.; Wang, X.; Carraway, H.; Sekeres, M.A.; Sukkur, A.; et al. Targeted therapy with the mutant IDH2 inhibitor enasidenib for high-risk IDH2-mutant myelodysplastic syndrome. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.N.; Pigneux, A.; Montesinos, P.; Wang, E.S.; et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: Phase 1 results of a phase 1/2 trial. Lancet Haematol. 2023, 10, e46–e58. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef]
- Rinke, J.; Müller, J.P.; Blaess, M.F.; Chase, A.; Meggendorfer, M.; Schäfer, V.; Winkelmann, N.; Haferlach, C.; Cross, N.C.P.; Hochhaus, A.; et al. Molecular characterization of EZH2 mutant patients with myelodysplastic/myeloproliferative neoplasms. Leukemia 2017, 31, 1936–1943. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a Prognostic Model and the Impact of Mutations in Patients With Lower-Risk Myelodysplastic Syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [Green Version]
- Lie, E.; Owen, C. Familial myelodysplastic syndromes: A review of the literature. Haematologica 2011, 96, 1536–1542. [Google Scholar] [CrossRef]
- Stengel, A.; Kern, W.; Meggendorfer, M.; Haferlach, T.; Haferlach, C. RUNX1 mutations in MDS, s-AML, and de novo AML: Differences in accompanying genetic alterations and outcome. Leuk. Lymphoma 2019, 60, 1334–1336. [Google Scholar] [CrossRef]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuhadra, N.; Al-Issa, K.; Mukherjee, S.; Hirsch, C.M.; Gerds, A.T.; Jha, B.K.; Adema, V.; Awada, H.; Asad, M.F.B.; Goyal, A.; et al. BCOR Mutations in Myelodysplastic Syndromes (MDS): Mutation Characteristics Impact Clinical Outcomes. Blood 2017, 130, 5304. [Google Scholar]
- Damm, F.; Chesnais, V.; Nagata, Y.; Yoshida, K.; Scourzic, L.; Okuno, Y.; Itzykson, R.; Sanada, M.; Shiraishi, Y.; Gelsi-Boyer, V.; et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood 2013, 122, 3169–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; Van Den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, S.; Haering, C.; Nasmyth, K. Chromosomal Cohesin Forms a Ring. Cell 2003, 112, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.-A.; Tsai, C.-H.; Lin, C.-C.; Chou, W.-C.; Kuo, Y.-Y.; Liu, C.-Y.; Tseng, M.-H.; Peng, Y.-L.; Liu, M.-C.; Liu, C.-W.; et al. Incorporation of mutations in five genes in the revised International Prognostic Scoring System can improve risk stratification in the patients with myelodysplastic syndrome. Blood Cancer J. 2018, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Sanada, M.; Suzuki, T.; Shih, L.-Y.; Otsu, M.; Kato, M.; Yamazaki, S.; Tamura, A.; Honda, H.; Sakata-Yanagimoto, M.; Kumano, K.; et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009, 460, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Hosono, N.; Makishima, H.; Mahfouz, R.; Przychodzen, B.; Yoshida, K.; Jerez, A.; LaFramboise, T.; Polprasert, C.; Clemente, M.J.; Shiraishi, Y.; et al. Recurrent genetic defects on chromosome 5q in myeloid neoplasms. Oncotarget 2017, 8, 6483–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, O.K.; Siddon, A.J.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Cin, P.D.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Palomo, L.; Meggendorfer, M.; Hutter, S.; Twardziok, S.; Ademà, V.; Fuhrmann, I.; Fuster-Tormo, F.; Xicoy, B.; Zamora, L.; Acha, P.; et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood 2020, 136, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graubert, T.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2012, 44, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Douet-Guilbert, N.; Soubise, B.; Bernard, D.G.; Troadec, M.-B. Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process. Diagnostics 2022, 12, 1658. [Google Scholar] [CrossRef]
- Wang, N.; Wang, F.; Shan, N.; Sui, X.; Xu, H. IDH1 Mutation Is an Independent Inferior Prognostic Indicator for Patients with Myelodysplastic Syndromes. Acta Haematol. 2017, 138, 143–151. [Google Scholar] [CrossRef]
- Thol, F.; Weissinger, E.M.; Krauter, J.; Wagner, K.; Damm, F.; Wichmann, M.; Gohring, G.; Schumann, C.; Bug, G.; Ottmann, O.; et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica 2010, 95, 1668–1674. [Google Scholar] [CrossRef] [Green Version]
- Maurya, N.; Mohanty, P.; Dhangar, S.; Panchal, P.; Jijina, F.; Mathan, S.L.P.; Shanmukhaiah, C.; Madkaikar, M.; Vundinti, B.R. Comprehensive analysis of genetic factors predicting overall survival in Myelodysplastic syndromes. Sci. Rep. 2022, 12, 5925. [Google Scholar] [CrossRef]
- Tamaki, H.; Ogawa, H.; Ohyashiki, K.; Ohyashiki, J.; Iwama, H.; Inoue, K.; Soma, T.; Oka, Y.; Tatekawa, T.; Oji, Y.; et al. The Wilms’ tumor gene WT1 is a good marker for diagnosis of disease progression of myelodysplastic syndromes. Leukemia 1999, 13, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Nagata, Y.; Makishima, H.; Sanada, M.; Shiozawa, Y.; Kon, A.; Yoshizato, T.; Sato-Otsubo, A.; Kataoka, K.; Shiraishi, Y.; et al. Somatic PHF6 mutations in 1760 cases with various myeloid neoplasms. Leukemia 2016, 30, 2270–2273. [Google Scholar] [CrossRef] [PubMed]

| Morphologically Defined | Genetically Defined | WHO 2022 | ICC 2022 |
|---|---|---|---|
| Ring sideroblasts | MDS with low blasts and SF3B1 mutation. Detection of ≥15% ring sideroblasts may substitute for SF3B1 mutation. | None | |
| Number of dysplastic lineages | Number of dysplastic lineages is no longer included in WHO 2022. | MDS, NOS without dysplasia (−7/del(7q) or complex, and any mutations except multihit TP53 or SF3B1 ≥ 10% VAF) | |
| MDS, NOS with single lineage dysplasia (any cytogenetics, except for MDS-de(5q), and any mutations except multihit TP53 and not meeting criteria MDS SF3B1) | |||
| MDS, NOS with multilineage dysplasia (any cytogenetics, except for MDS-de(5q), and any mutations except multi-hit TP53 and not meeting criteria MDS SF3B1) | |||
| Blasts% | MDS with low blasts (MDS-LB): <5% bone marrow (BM) and <2% peripheral blood (PB). | None | |
| MDS, hypoplastic (MDS-h): <25% BM cellularity, age-adjusted | None | ||
| MDS with increased blasts (MDS-IB): MDS-IB1: 5–9% BM or 2–9% PB MDS-IB2: 10–19% BM or 5–19% PB or Auer rods | MDS with excess blasts (MDS-EB): 5–9% BM or 2–9% PB, any cytogenetics or mutations, except multihit TP53. | ||
| MDS with fibrosis (MDS-f): 5–19% BM or 2–19% PB | MDS/AML: 10–19% BM or PB blasts with any cytogenetics, except for AML-defining, and any mutations except for NPM1, bZIP CEBPA, and TP53 | ||
| Genetically defined Subtypes | Isolated 5q | MDS with low blasts and isolated 5q deletion (MDS-5q): 5q deletion alone or with 1 other abnormality other than monosomy 7 or 7q deletion | MDS with del(5q): del(5q) with up to one additional, except for −7/del(7q), with any mutations except multihit TP53 |
| SF3B1 | MDS with low blasts and SF3B1: mutation in the absence of 5q deletion, monosomy 7, or complex karyotype | MDS with mutated SF3B1: SF3B1 (≥10% VAF) without multihit TP53 or RUNX1, with any cytogenetics except for isolated del(5q), −7/del(7q), abn3q26.2, or complex. | |
| TP53 | MDS with biallelic TP53 inactivation (MDS-biTP53): two or more TP53 mutations or 1 mutation with evidence of TP53 copy number loss or copy neutral loss of heterozygosity. In the presence of ≤20% BM or PB blasts | Myeloid neoplasm with mutated TP53 (MDS-TP53, MDS/AML-TP53) Defined as 2 distinct TP53 mutations (each VAF > 10%) OR a single TP53 mutation with (1) 17p deletion on cytogenetics; (2) VAF of >50%; or (3) copy-neutral LOH at the 17p TP53 locus. |
| Score | |||||||
|---|---|---|---|---|---|---|---|
| Variable | 0 | 0.5 | 1.0 | 1.5 | 2.0 | 3.0 | 4.0 |
| IPSS | |||||||
| Bone marrow blast (%) | <5% | 5–10% | 11–20% | 21–30% | |||
| Karyotype † | Good | Intermediate | Poor | ||||
| Cytopenias †† | 0/1 | 2/3 | |||||
| IPSS-R | |||||||
| Cytogenetics ††† | Very good | Good | Intermediate | Poor | Very poor | ||
| Bone marrow blast (%) | ≤2% | >2 to <5% | 5–10% | >10% | |||
| Hemoglobin (g/dL) | ≥10 | 8 to <10 | <8 | ||||
| Platelets (cells/µL) | ≥100 | 50–100 | <50 | ||||
| Absolute neutrophil count (cell/µL) | ≥0.8 | <0.8 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoff, F.W.; Madanat, Y.F. Molecular Drivers of Myelodysplastic Neoplasms (MDS)—Classification and Prognostic Relevance. Cells 2023, 12, 627. https://doi.org/10.3390/cells12040627
Hoff FW, Madanat YF. Molecular Drivers of Myelodysplastic Neoplasms (MDS)—Classification and Prognostic Relevance. Cells. 2023; 12(4):627. https://doi.org/10.3390/cells12040627
Chicago/Turabian StyleHoff, Fieke W., and Yazan F. Madanat. 2023. "Molecular Drivers of Myelodysplastic Neoplasms (MDS)—Classification and Prognostic Relevance" Cells 12, no. 4: 627. https://doi.org/10.3390/cells12040627