
Alzheimer’s Disease-Associated Alternative Splicing of CD33 Is Regulated by the HNRNPA Family Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. Cellular Splicing Assay
2.4. SDS-PAGE and Western Blotting
2.5. Ribonucleoprotein Immunoprecipitation (RIP)
2.6. Statistical Analysis
2.7. Animals
3. Results
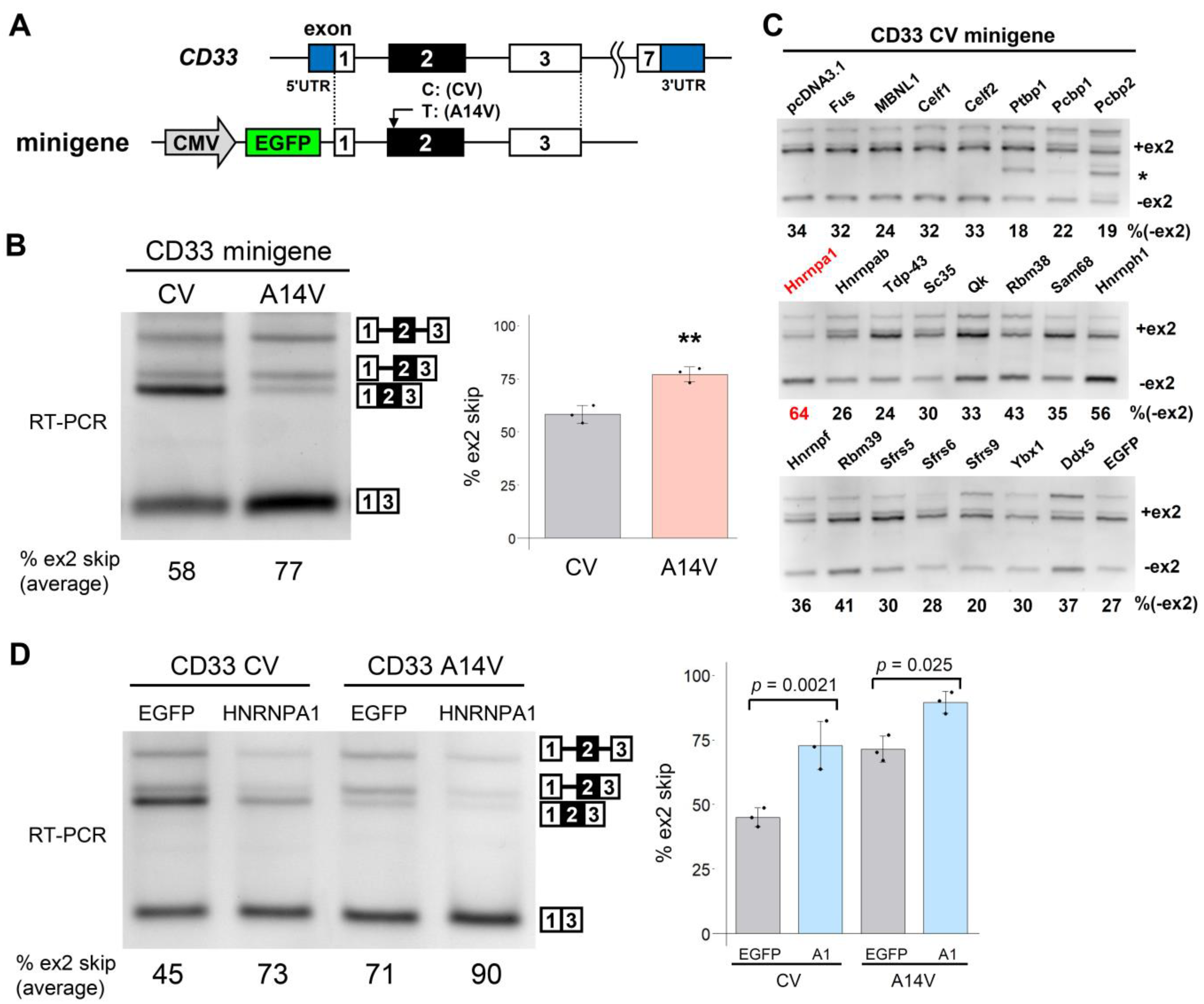
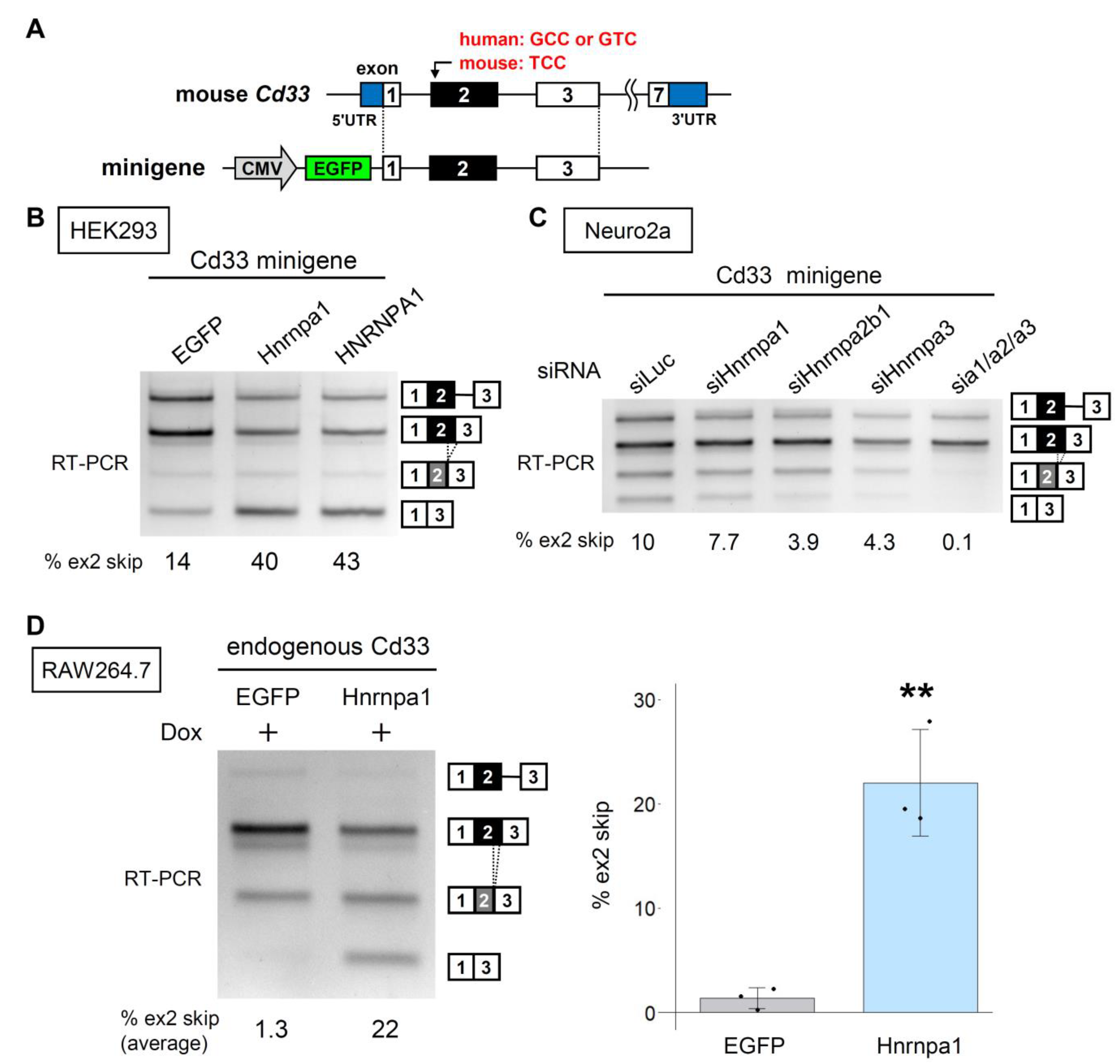
3.1. Screening of RBPs for the Regulators of CD33 Exon 2 Splicing
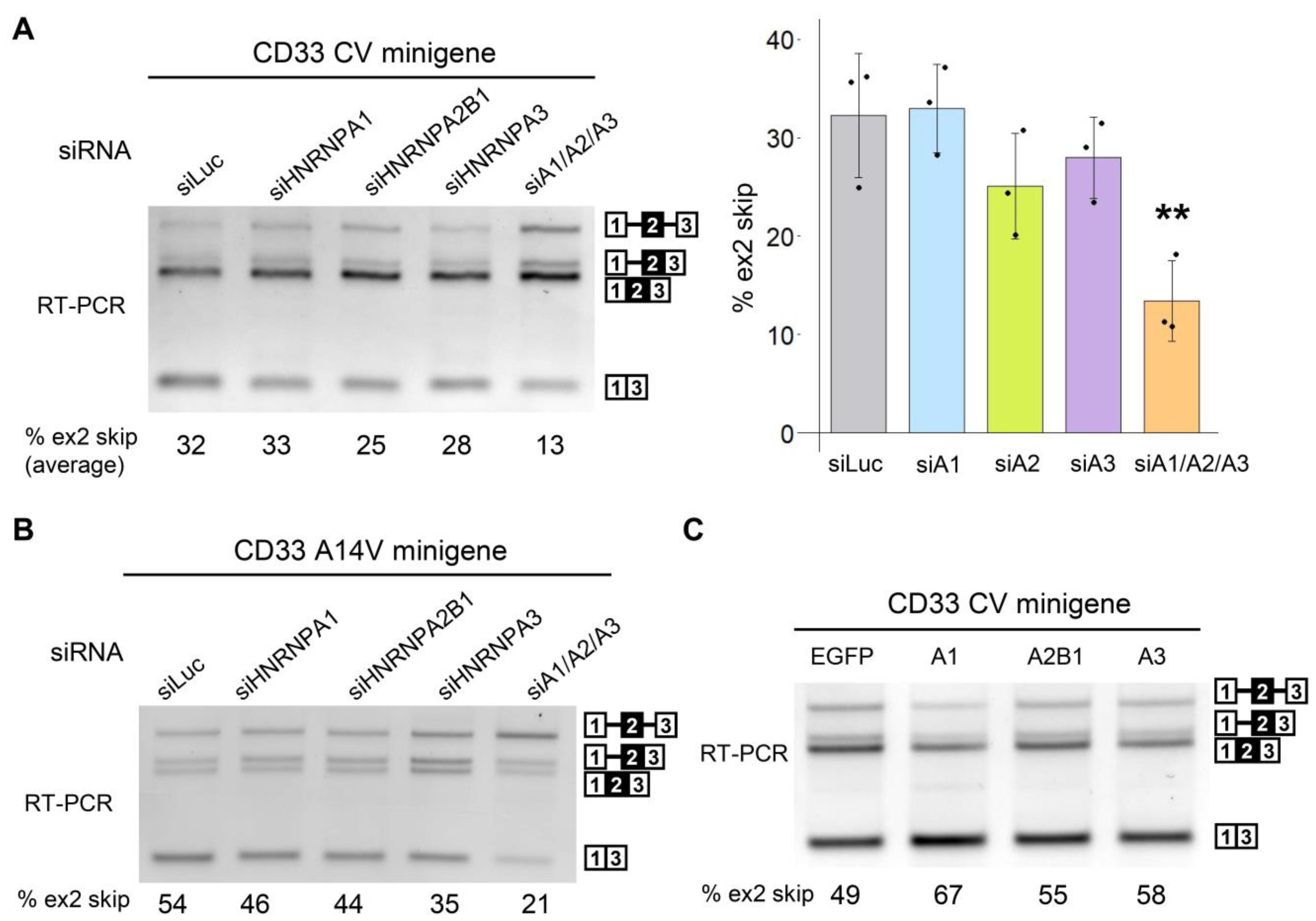
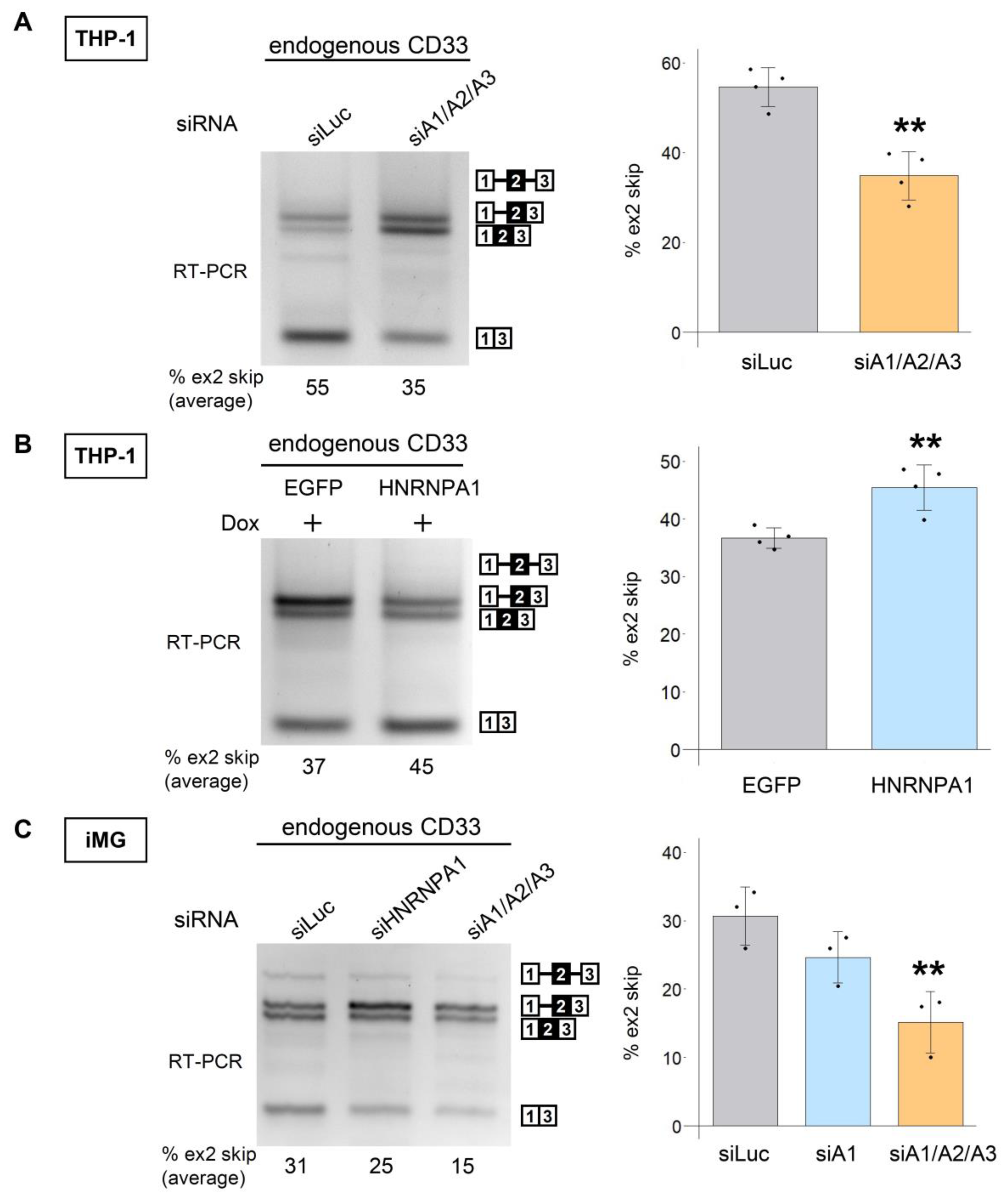
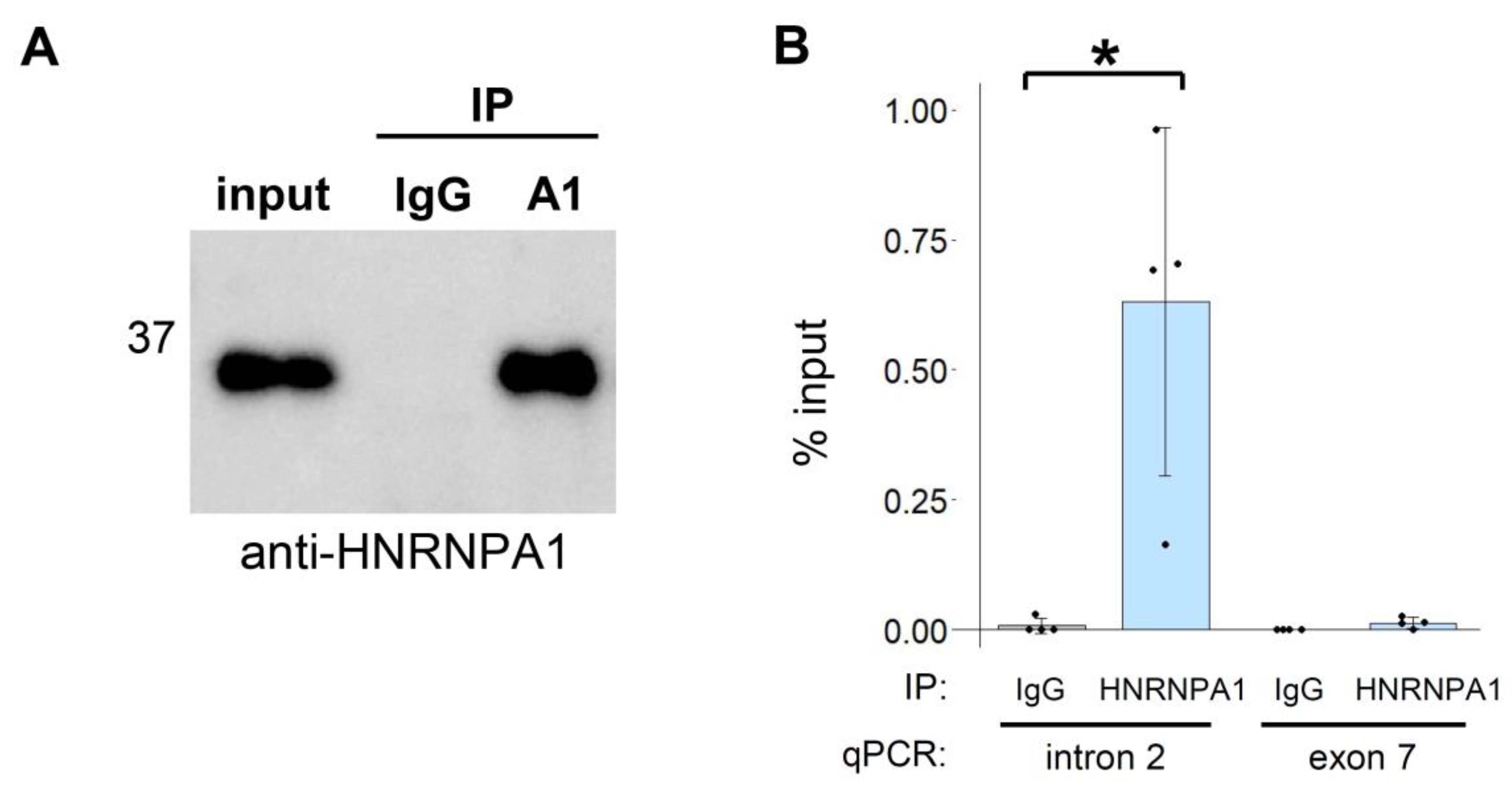
3.2. The HNRNPA Family Proteins Redundantly Regulate the Splicing of CD33 Exon 2
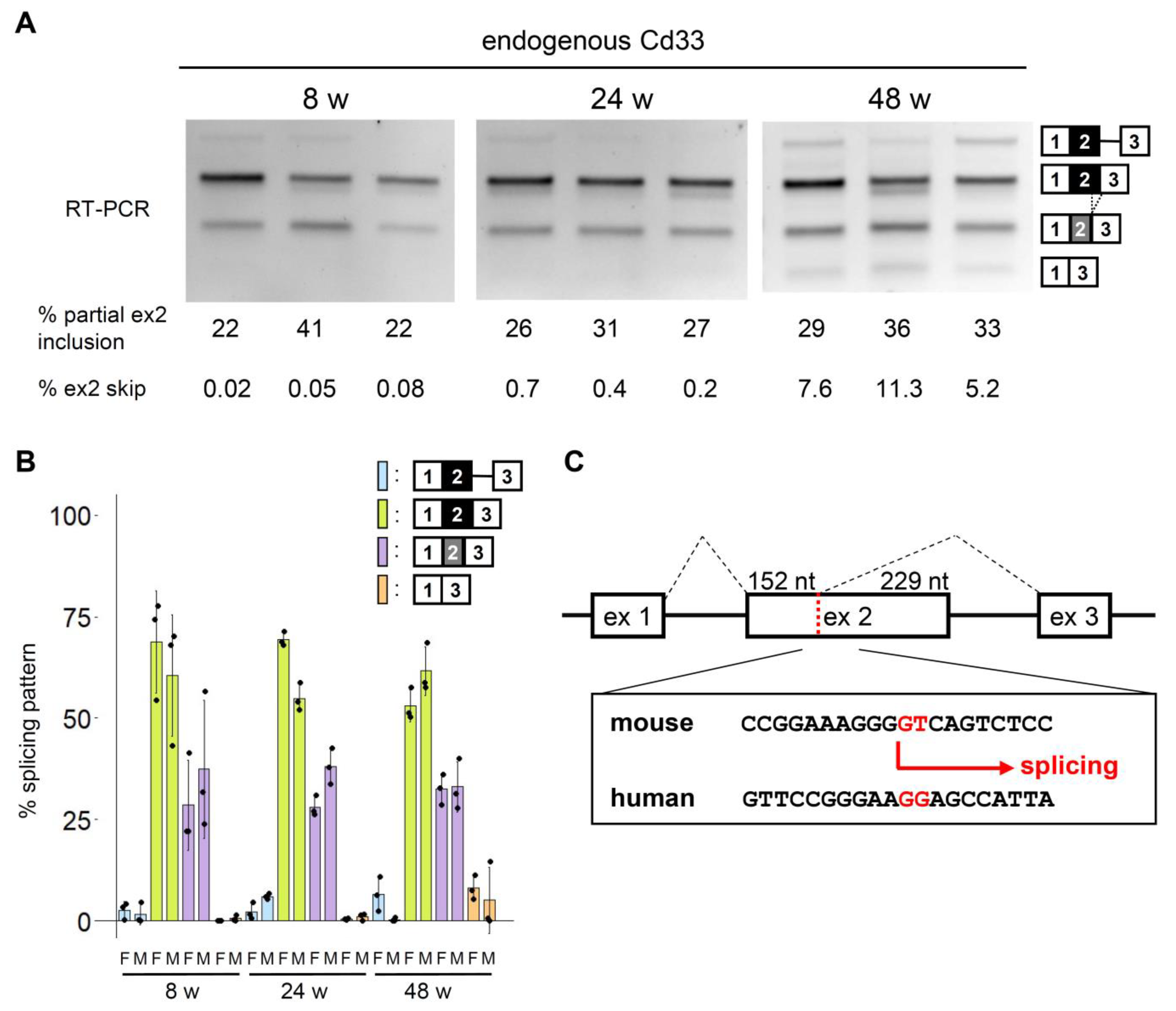
3.3. Conserved and Unique Features of Mouse Cd33 Splicing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duyckaerts, C.; Delatour, B.; Potier, M.-C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Raybould, R.; Sims, R. Searching the Dark Genome for Alzheimer’s Disease Risk Variants. Brain Sci. 2021, 11, 332. [Google Scholar] [CrossRef]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Ulvestad, E.; Waage, A.; Antel, J.P.; McLaurin, J. Activation of adult human derived microglia by myelin phagocytosis in vitro. J. Neurosci. Res. 1994, 38, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.D.; Kelm, S.; Barber, E.K.; Crocker, P. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood 1995, 85, 2005–2012. [Google Scholar] [CrossRef]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef]
- Paul, S.P.; Taylor, L.S.; Stansbury, E.K.; McVicar, D.W. Myeloid specific human CD33 is an inhibitory receptor with differential ITIM function in recruiting the phosphatases SHP-1 and SHP-2. Blood 2000, 96, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835.e7. [Google Scholar] [CrossRef]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef]
- Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 2014, 23, 2729–2736. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Abeta1-42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.A.; Thornton, P.; Brown, G.C. CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease-protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion. J. Neurochem. 2021, 158, 297–310. [Google Scholar] [CrossRef]
- Wissfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Chappie, T.A.; Abdelmessih, M.; Ambroise, C.W.; Boehm, M.; Cai, M.; Green, M.; Guilmette, E.; Steppan, C.M.; Stevens, L.M.; Wei, L.; et al. Discovery of Small-Molecule CD33 Pre-mRNA Splicing Modulators. ACS Med. Chem. Lett. 2022, 13, 55–62. [Google Scholar] [CrossRef]
- van Bergeijk, P.; Seneviratne, U.; Aparicio-Prat, E.; Stanton, R.; Hasson, S.A. SRSF1 and PTBP1 Are trans-Acting Factors That Suppress the Formation of a CD33 Splicing Isoform Linked to Alzheimer’s Disease Risk. Mol. Cell Biol. 2019, 39, e00568-18. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Oma, Y.; Hattori, N.; Ishiura, S.; Nukina, N. Nuclear localization of MBNL1: Splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum. Mol. Genet. 2015, 24, 740–756. [Google Scholar] [CrossRef] [PubMed]
- Yanaizu, M.; Washizu, C.; Nukina, N.; Satoh, J.-I.; Kino, Y. CELF2 regulates the species-specific alternative splicing of TREM2. Sci. Rep. 2020, 10, 17995. [Google Scholar] [CrossRef]
- Yanaizu, M.; Sakai, K.; Tosaki, Y.; Kino, Y.; Satoh, J.-I. Small nuclear RNA-mediated modulation of splicing reveals a therapeutic strategy for a TREM2 mutation and its post-transcriptional regulation. Sci. Rep. 2018, 8, 6937. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. One Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Malik, M.; Chiles, J., III; Xi, H.S.; Medway, C.; Simpson, J.; Potluri, S.; Howard, D.; Liang, Y.; Paumi, C.M.; Mukherjee, S. Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum. Mol. Genet. 2015, 24, 3557–3570. [Google Scholar] [CrossRef]
- Roy, R.; Huang, Y.; Seckl, M.J.; Pardo, O.E. Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip. Rev. RNA 2017, 8, e1431. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, S.L. The roles of hnRNP A2/B1 in RNA biology and disease. Wiley Interdiscip. Rev. RNA 2021, 12, e1612. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lin, W.; Zhang, C.; Ash, P.E.; Verma, M.; Kwan, J.; van Vliet, E.; Yang, Z.; Cruz, A.L.; Boudeau, S.; et al. Interaction of tau with HNRNPA2B1 and N(6)-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 2021, 81, 4209–4227.e12. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.E.; Hutchinson, C.; Popescu, B.F.; Levin, M.C. Neuronal RNA-binding protein dysfunction in multiple sclerosis cortex. Ann. Clin. Transl. Neurol. 2020, 7, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kim, D.; Choi, N.; Oh, J.; Ha, J.; Zhou, J.; Zheng, X.; Shen, H. hnRNP A1 Regulates Alternative Splicing of Tau Exon 10 by Targeting 3’ Splice Sites. Cells 2020, 9, 936. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Rodrigues, E.; Jung, J.; Luzentales-Simpson, M.; Enterina, J.R.; Galleguillos, D.; Laurent, C.D.S.; Nakhaei-Nejad, M.; Fuchsberger, F.F.; Streith, L.; et al. Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun. Biol. 2019, 2, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komuro, R.; Honda, Y.; Yanaizu, M.; Nagahama, M.; Kino, Y. Alzheimer’s Disease-Associated Alternative Splicing of CD33 Is Regulated by the HNRNPA Family Proteins. Cells 2023, 12, 602. https://doi.org/10.3390/cells12040602
Komuro R, Honda Y, Yanaizu M, Nagahama M, Kino Y. Alzheimer’s Disease-Associated Alternative Splicing of CD33 Is Regulated by the HNRNPA Family Proteins. Cells. 2023; 12(4):602. https://doi.org/10.3390/cells12040602
Chicago/Turabian StyleKomuro, Riho, Yuka Honda, Motoaki Yanaizu, Masami Nagahama, and Yoshihiro Kino. 2023. "Alzheimer’s Disease-Associated Alternative Splicing of CD33 Is Regulated by the HNRNPA Family Proteins" Cells 12, no. 4: 602. https://doi.org/10.3390/cells12040602