
Ceramide Nanoliposomes as Potential Therapeutic Reagents for Asthma
 , ,
, ,
Abstract
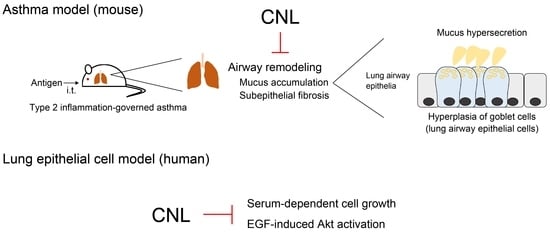
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. CNL Preparation
2.3. Sensitization and Challenges for Asthmatic Model
2.4. Ethics Statement
2.5. Bronchoalveolar Lavage (BAL) Fluid Collection
2.6. Immunohistochemistry
2.7. Lung Histology
2.8. ELISA
2.9. Analysis of ILC2, Treg, and Tr1 Cells
2.10. Cell Culture
2.11. Quantitative Real-Time PCR
2.12. Cell Viability Assay
2.13. Immunoblotting
2.14. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Prim. 2015, 1, 15025. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Vandivier, R.W.; Tuder, R.M. Vascular endothelial growth factor in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L209–L221. [Google Scholar] [CrossRef]
- Chan, R.; Stewart, K.; Misirovs, R.; Lipworth, B.J. Targeting Downstream Type 2 Cytokines or Upstream Epithelial Alarmins for Severe Asthma. J. Allergy Clin. Immunol. Pract. 2022, 10, 1497–1505. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef]
- Davis, D.; Kannan, M.; Wattenberg, B. Orm/ORMDL proteins: Gate guardians and master regulators. Adv. Biol. Regul. 2018, 70, 3–18. [Google Scholar] [CrossRef]
- Ono, J.G.; Kim, B.I.; Zhao, Y.; Christos, P.J.; Tesfaigzi, Y.; Worgall, T.S.; Worgall, S. Decreased sphingolipid synthesis in children with 17q21 asthma-risk genotypes. J. Clin. Investig. 2020, 130, 921–926. [Google Scholar] [CrossRef]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Futerman, A.H.; Riezman, H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 2008, 20, 1010–1018. [Google Scholar] [CrossRef]
- Kitatani, K.; Akiba, S.; Hayama, M.; Sato, T. Ceramide accelerates dephosphorylation of extracellular signal-regulated kinase 1/2 to decrease prostaglandin D(2) production in RBL-2H3 cells. Arch. Biochem Biophys 2001, 395, 208–214. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Bielawski, J.; Taha, T.A.; Jenkins, R.W.; Senkal, C.E.; Ogretmen, B.; Obeid, L.M.; Hannun, Y.A. Protein kinase C-induced activation of a ceramide/protein phosphatase 1 pathway leading to dephosphorylation of p38 MAPK. J. Biol. Chem. 2006, 281, 36793–36802. [Google Scholar] [CrossRef]
- Kitatani, K.; Sheldon, K.; Anelli, V.; Jenkins, R.W.; Sun, Y.; Grabowski, G.A.; Obeid, L.M.; Hannun, Y.A. Acid beta-glucosidase 1 counteracts p38delta-dependent induction of interleukin-6: Possible role for ceramide as an anti-inflammatory lipid. J. Biol. Chem. 2009, 284, 12979–12988. [Google Scholar] [CrossRef]
- Nakamura, Y.; Nakashima, S.; Ojio, K.; Banno, Y.; Miyata, H.; Nozawa, Y. Ceramide inhibits IgE-mediated activation of phospholipase D, but not of phospholipase C, in rat basophilic leukemia (RBL-2H3) cells. J. Immunol 1996, 156, 256–262. [Google Scholar] [CrossRef]
- Izawa, K.; Yamanishi, Y.; Maehara, A.; Takahashi, M.; Isobe, M.; Ito, S.; Kaitani, A.; Matsukawa, T.; Matsuoka, T.; Nakahara, F.; et al. The receptor LMIR3 negatively regulates mast cell activation and allergic responses by binding to extracellular ceramide. Immunity 2012, 37, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Fox, T.; Adhikary, G.; Kester, M.; Pearlman, E. Inhibition of corneal inflammation by liposomal delivery of short-chain, C-6 ceramide. J. Leukoc Biol 2008, 83, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Tagaram, H.R.; Divittore, N.A.; Barth, B.M.; Kaiser, J.M.; Avella, D.; Kimchi, E.T.; Jiang, Y.; Isom, H.C.; Kester, M.; Staveley-O’Carroll, K.F. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut 2011, 60, 695–701. [Google Scholar] [CrossRef]
- Kester, M.; Bassler, J.; Fox, T.E.; Carter, C.J.; Davidson, J.A.; Parette, M.R. Preclinical development of a C6-ceramide NanoLiposome, a novel sphingolipid therapeutic. Biol. Chem. 2015, 396, 737–747. [Google Scholar] [CrossRef]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide Nanoliposomes as a MLKL-Dependent, Necroptosis-Inducing, Chemotherapeutic Reagent in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef]
- Barth, B.M.; Wang, W.; Toran, P.T.; Fox, T.E.; Annageldiyev, C.; Ondrasik, R.M.; Keasey, N.R.; Brown, T.J.; Devine, V.G.; Sullivan, E.C.; et al. Sphingolipid metabolism determines the therapeutic efficacy of nanoliposomal ceramide in acute myeloid leukemia. Blood Adv. 2019, 3, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Comer, B.S.; Ba, M.; Singer, C.A.; Gerthoffer, W.T. Epigenetic targets for novel therapies of lung diseases. Pharmacol. Ther. 2015, 147, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Tanaka, Y.; Shimora, H.; Takemoto, N.; Nomura, M.; Terakawa, R.; Hashimoto, K.; Sakae, H.; Kanda, A.; Iwai, H.; et al. Pathogenic changes in group 2 innate lymphoid cells (ILC2s) in a steroid-insensitive asthma model of mice. Eur. J. Pharmacol. 2022, 916, 174732. [Google Scholar] [CrossRef]
- Ho, W.; Furst, A. Intratracheal instillation method for mouse lungs. Oncology 1973, 27, 385–393. [Google Scholar] [CrossRef]
- Nabe, T.; Zindl, C.L.; Jung, Y.W.; Stephens, R.; Sakamoto, A.; Kohno, S.; Atkinson, T.P.; Chaplin, D.D. Induction of a late asthmatic response associated with airway inflammation in mice. Eur. J. Pharmacol. 2005, 521, 144–155. [Google Scholar] [CrossRef]
- Matsuda, M.; Tabuchi, Y.; Nishimura, K.; Nakamura, Y.; Sekioka, T.; Kadode, M.; Kawabata, K.; Nabe, T. Increased expression of CysLT2 receptors in the lung of asthmatic mice and role in allergic responses. Prostaglandins Leukot. Essent. Fatty Acids 2018, 131, 24–31. [Google Scholar] [CrossRef]
- Matsuda, M.; Doi, K.; Tsutsumi, T.; Inaba, M.; Hamaguchi, J.; Terada, T.; Kawata, R.; Kitatani, K.; Nabe, T. Adoptive transfer of type 1 regulatory T cells suppressed the development of airway hyperresponsiveness in ovalbumin-induced airway inflammation model mice. J. Pharmacol. Sci. 2019, 141, 139–145. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Chatila, T.A. Regulatory T cells in allergic diseases. J. Allergy Clin. Immunol. 2016, 138, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Gregori, S.; Passerini, L.; Roncarolo, M.G. Clinical Outlook for Type-1 and FOXP3(+) T Regulatory Cell-Based Therapy. Front. Immunol. 2015, 6, 593. [Google Scholar] [CrossRef]
- Klein Wolterink, R.G.; Kleinjan, A.; van Nimwegen, M.; Bergen, I.; de Bruijn, M.; Levani, Y.; Hendriks, R.W. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol. 2012, 42, 1106–1116. [Google Scholar] [CrossRef]
- Mjosberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; te Velde, A.A.; Fokkens, W.J.; van Drunen, C.M.; Spits, H. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Peebles, R.S., Jr.; Aronica, M.A. Proinflammatory Pathways in the Pathogenesis of Asthma. Clin. Chest. Med. 2019, 40, 29–50. [Google Scholar] [CrossRef]
- Hattrup, C.L.; Gendler, S.J. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 2008, 70, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Korfhagen, T.R.; Bruno, M.D.; Kitzmiller, J.A.; Wan, H.; Wert, S.E.; Khurana Hershey, G.K.; Chen, G.; Whitsett, J.A. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J. Clin. Invest. 2007, 117, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Gras, D.; Chanez, P.; Vachier, I.; Petit, A.; Bourdin, A. Bronchial epithelium as a target for innovative treatments in asthma. Pharmacol Ther 2013, 140, 290–305. [Google Scholar] [CrossRef]
- Ma, J.; Rubin, B.K.; Voynow, J.A. Mucins, Mucus, and Goblet Cells. Chest 2018, 154, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A. Functions of ceramide in coordinating cellular responses to stress. Science 1996, 274, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Kronke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Schady, D.; Usta, J.; Wood, R.; Kraveka, J.M.; Luberto, C.; Birbes, H.; Hannun, Y.A.; Obeid, L.M. Role of ceramide in mediating the inhibition of telomerase activity in A549 human lung adenocarcinoma cells. J. Biol. Chem. 2001, 276, 24901–24910. [Google Scholar] [CrossRef] [PubMed]
- Shatos, M.A.; Hodges, R.R.; Oshi, Y.; Bair, J.A.; Zoukhri, D.; Kublin, C.; Lashkari, K.; Dartt, D.A. Role of cPKCalpha and nPKCepsilon in EGF-stimulated goblet cell proliferation. Invest. Ophthalmol. Vis. Sci. 2009, 50, 614–620. [Google Scholar] [CrossRef]
- Le Cras, T.D.; Acciani, T.H.; Mushaben, E.M.; Kramer, E.L.; Pastura, P.A.; Hardie, W.D.; Korfhagen, T.R.; Sivaprasad, U.; Ericksen, M.; Gibson, A.M.; et al. Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L414–L421. [Google Scholar] [CrossRef]
- Adcock, I.M.; Caramori, G.; Chung, K.F. New targets for drug development in asthma. Lancet 2008, 372, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.F. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest 2011, 139, 1470–1479. [Google Scholar] [CrossRef]
- Khorasanizadeh, M.; Eskian, M.; Gelfand, E.W.; Rezaei, N. Mitogen-activated protein kinases as therapeutic targets for asthma. Pharmacol. Ther. 2017, 174, 112–126. [Google Scholar] [CrossRef]
- Duan, W.; Chan, J.H.; McKay, K.; Crosby, J.R.; Choo, H.H.; Leung, B.P.; Karras, J.G.; Wong, W.S. Inhaled p38alpha mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am. J. Respir. Crit. Care Med. 2005, 171, 571–578. [Google Scholar] [CrossRef]
- Nath, P.; Leung, S.Y.; Williams, A.; Noble, A.; Chakravarty, S.D.; Luedtke, G.R.; Medicherla, S.; Higgins, L.S.; Protter, A.; Chung, K.F. Importance of p38 mitogen-activated protein kinase pathway in allergic airway remodelling and bronchial hyperresponsiveness. Eur. J. Pharmacol. 2006, 544, 160–167. [Google Scholar] [CrossRef]
- Atherton, H.C.; Jones, G.; Danahay, H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L730–L739. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Rosenthal, P.; Beppu, A.; Mueller, J.L.; Hoffman, H.M.; Tam, A.B.; Doherty, T.A.; McGeough, M.D.; Pena, C.A.; Suzukawa, M.; et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J. Immunol. 2014, 192, 3475–3487. [Google Scholar] [CrossRef]
- Petrache, I.; Kamocki, K.; Poirier, C.; Pewzner-Jung, Y.; Laviad, E.L.; Schweitzer, K.S.; Van Demark, M.; Justice, M.J.; Hubbard, W.C.; Futerman, A.H. Ceramide synthases expression and role of ceramide synthase-2 in the lung: Insight from human lung cells and mouse models. PLoS ONE 2013, 8, e62968. [Google Scholar] [CrossRef] [PubMed]
- James, B.N.; Oyeniran, C.; Sturgill, J.L.; Newton, J.; Martin, R.K.; Bieberich, E.; Weigel, C.; Maczis, M.A.; Palladino, E.N.D.; Lownik, J.C.; et al. Ceramide in apoptosis and oxidative stress in allergic inflammation and asthma. J. Allergy Clin. Immunol. 2021, 147, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Roviezzo, F.; Di Lorenzo, A.; Bucci, M.; Brancaleone, V.; Vellecco, V.; De Nardo, M.; Orlotti, D.; De Palma, R.; Rossi, F.; D’Agostino, B.; et al. Sphingosine-1-phosphate/sphingosine kinase pathway is involved in mouse airway hyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 2007, 36, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Price, M.M.; Oskeritzian, C.A.; Falanga, Y.T.; Harikumar, K.B.; Allegood, J.C.; Alvarez, S.E.; Conrad, D.; Ryan, J.J.; Milstien, S.; Spiegel, S. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J. Allergy Clin. Immunol. 2013, 131, 501–511. [Google Scholar] [CrossRef]
- Riemma, M.A.; Cerqua, I.; Romano, B.; Irollo, E.; Bertolino, A.; Camerlingo, R.; Granato, E.; Rea, G.; Scala, S.; Terlizzi, M.; et al. Sphingosine-1-phosphate/TGF-beta axis drives epithelial mesenchymal transition in asthma-like disease. Br. J. Pharmacol. 2022, 179, 1753–1768. [Google Scholar] [CrossRef]
- Rogers, D.F. Airway goblet cells: Responsive and adaptable front-line defenders. Eur. Respir. J. 1994, 7, 1690–1706. [Google Scholar] [CrossRef]
- Rose, M.C.; Voynow, J.A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 2006, 86, 245–278. [Google Scholar] [CrossRef] [Green Version]
- Curran, D.R.; Cohn, L. Advances in mucous cell metaplasia: A plug for mucus as a therapeutic focus in chronic airway disease. Am. J. Respir. Cell Mol. Biol. 2010, 42, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Rajavelu, P.; Chen, G.; Xu, Y.; Kitzmiller, J.A.; Korfhagen, T.R.; Whitsett, J.A. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J. Clin. Invest. 2015, 125, 2021–2031. [Google Scholar] [CrossRef]
- Salli, U.; Fox, T.E.; Carkaci-Salli, N.; Sharma, A.; Robertson, G.P.; Kester, M.; Vrana, K.E. Propagation of undifferentiated human embryonic stem cells with nano-liposomal ceramide. Stem Cells Dev. 2009, 18, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia 2001, 15, 1153–1160. [Google Scholar] [CrossRef]
- Coant, N.; Garcia-Barros, M.; Zhang, Q.; Obeid, L.M.; Hannun, Y.A. AKT as a key target for growth promoting functions of neutral ceramidase in colon cancer cells. Oncogene 2018, 37, 3852–3863. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Lucic, I.; Rathinaswamy, M.K.; Truebestein, L.; Hamelin, D.J.; Burke, J.E.; Leonard, T.A. Conformational sampling of membranes by Akt controls its activation and inactivation. Proc. Natl. Acad Sci. USA 2018, 115, E3940–E3949. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Kitatani, K.; Usui, T.; Sriraman, S.K.; Toyoshima, M.; Ishibashi, M.; Shigeta, S.; Nagase, S.; Sakamoto, M.; Ogiso, H.; Okazaki, T.; et al. Ceramide limits phosphatidylinositol-3-kinase C2beta-controlled cell motility in ovarian cancer: Potential of ceramide as a metastasis-suppressor lipid. Oncogene 2016, 35, 2801–2812. [Google Scholar] [CrossRef]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef]
- Bhavsar, P.; Hew, M.; Khorasani, N.; Torrego, A.; Barnes, P.J.; Adcock, I.; Chung, K.F. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax 2008, 63, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type (× 105 Cells/Lung) | No OVA Challenge | OVA Challenge + Ghost | OVA Challenge + CNL |
|---|---|---|---|
| ILC2 | 1.67 ± 0.21 (n = 6) | 7.13 ± 0.85 (n = 6) | 8.15 ± 0.81 (n = 4) |
| Treg | 1.25 ± 0.34 (n = 4) | 6.67 ± 2.69 (n = 4) | 3.74 ± 1.31 (n = 4) |
| Tr1 | 0.49 ± 0.10 (n = 4) | 5.74 ± 0.83 (n = 4) | 3.74 ± 0.80 (n = 4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakae, H.; Ogiso, Y.; Matsuda, M.; Shimora, H.; Deering, T.; Fox, T.E.; Kester, M.; Nabe, T.; Kitatani, K. Ceramide Nanoliposomes as Potential Therapeutic Reagents for Asthma. Cells 2023, 12, 591. https://doi.org/10.3390/cells12040591
Sakae H, Ogiso Y, Matsuda M, Shimora H, Deering T, Fox TE, Kester M, Nabe T, Kitatani K. Ceramide Nanoliposomes as Potential Therapeutic Reagents for Asthma. Cells. 2023; 12(4):591. https://doi.org/10.3390/cells12040591
Chicago/Turabian StyleSakae, Harumi, Yuri Ogiso, Masaya Matsuda, Hayato Shimora, Tye Deering, Todd E. Fox, Mark Kester, Takeshi Nabe, and Kazuyuki Kitatani. 2023. "Ceramide Nanoliposomes as Potential Therapeutic Reagents for Asthma" Cells 12, no. 4: 591. https://doi.org/10.3390/cells12040591