
Dysregulated Signaling at Postsynaptic Density: A Systematic Review and Translational Appraisal for the Pathophysiology, Clinics, and Antipsychotics’ Treatment of Schizophrenia

 , and
, and
Abstract
: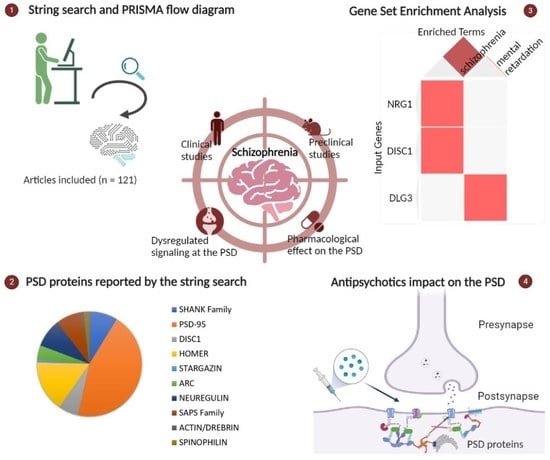
1. Introduction: The PSD Landscape, Receptors, Adaptors, and Scaffolding Proteins Linking to Schizophrenia
- How is PSD involvement coherent within the present framework of aberrant dopamine-glutamate interaction in schizophrenia?
- What is the contribution of each PSD protein associated with the pathophysiology of schizophrenia?
- How might innovative methodology and discovery contribute to a non-canonical conceptualization of PSD’s role in schizophrenia?
- Finally, how does antipsychotic treatment impact PSD proteins, and how might it be instrumental in developing new therapeutic strategies for schizophrenia?
2. Materials and Methods
3. The PSD Definition and Structure
3.1. The Spatial and Temporal Distribution of PSD
3.2. Glutamate Receptors: NMDAR and Beyond
4. The Shank Family and Schizophrenia: Overlapping with Other Neurodevelopmental Diseases
4.1. Shank Structure and Topography
4.2. Shank Connectome and Signalosome
4.3. Shank and Schizophrenia
4.4. Shank and Psychotropic Drugs
5. The PSD-95
5.1. PSD-95 Structure and Topography
5.2. PSD-95 Connectome and Signalosome
5.3. PSD-95 in Schizophrenia and Relevance for Antipsychotic Treatment
6. DISC1 a Scaffolding Protein
6.1. DISC1 Distribution, Structure and General Function
6.2. DISC1 Connectome and Signalosome
6.3. DISC1 and Schizophrenia: Human Common and Ultrarare DISC1 Variants
6.4. DISC1, a Drugable Molecule?
7. The Homer Proteins at the PSD
7.1. Homer1: Distribution, Structure, and General Function
7.2. The Homer Connectome and Signalosome
7.3. Homer, Schizophrenia, and Antipsychotic Treatment
8. Other PSD Proteins
8.1. The Role of PSD Transmembrane AMPAR Regulatory Proteins in Schizophrenia: Stargazin
8.2. Arc and PSD
8.2.1. Arc Distribution, Connectome, and Signalosome
8.2.2. Arc in Schizophrenia and Antipsychotic Modulation
8.3. Neuregulin
8.4. Synapse-Associated Proteins Family
8.4.1. Synapse-Associated Proteins Spatial and Temporal Distribution at the PSD
8.4.2. Synapse-Associated Proteins in Psychotic Disorders: Clinical and Pharmacological Implications
9. The PSD Interactome in Schizophrenia: Actin and Drebrin Interacting with the Main Scaffolding/Adaptor Protein and Receptors
9.1. Role of Actin Cytoskeleton in Dendritic Spine Morphogenesis and Implications in Schizophrenia
9.2. Drebrin in Synaptic Dysfunction and Neuropsychiatric Implications
9.3. Spinophilin Regulation in the Dynamic Morphological Changes of Dendritic Spine: Implications in Schizophrenia
10. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Bartolomeis, A.; Tomasetti, C. Calcium-dependent networks in dopamine-glutamate interaction: The role of postsynaptic scaffolding proteins. Mol. Neurobiol. 2012, 46, 275–296. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, H.; Li, C. SAPAP Scaffold Proteins: From Synaptic Function to Neuropsychiatric Disorders. Cells 2022, 11, 3815. [Google Scholar] [CrossRef] [PubMed]
- Cresto, N.; Lebrun, N.; Dumont, F.; Letourneur, F.; Billuart, P.; Rouach, N. Hippocampal Excitatory Synaptic Transmission and Plasticity Are Differentially Altered during Postnatal Development by Loss of the X-Linked Intellectual Disability Protein Oligophrenin-1. Cells 2022, 11, 1545. [Google Scholar] [CrossRef]
- Gentile, J.E.; Carrizales, M.G.; Koleske, A.J. Control of Synapse Structure and Function by Actin and Its Regulators. Cells 2022, 11, 603. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Shirai, Y.; Kouyama-Suzuki, E.; Zhou, M.; Yoshizawa, T.; Yanagawa, T.; Mori, T.; Tabuchi, K. IQSEC2 Deficiency Results in Abnormal Social Behaviors Relevant to Autism by Affecting Functions of Neural Circuits in the Medial Prefrontal Cortex. Cells 2021, 10, 2724. [Google Scholar] [CrossRef] [PubMed]
- Ly, S.; Strus, E.; Naidoo, N. Genetic disruption of the putative binding site for Homer on DmGluRA reduces sleep in Drosophila. Sleep 2020, 43, 190. [Google Scholar] [CrossRef]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, e1166. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Barone, A.; Buonaguro, E.F.; Tomasetti, C.; Vellucci, L.; Iasevoli, F. The Homer1 family of proteins at the crossroad of dopamine-glutamate signaling: An emerging molecular “Lego” in the pathophysiology of psychiatric disorders. A systematic review and translational insight. Neurosci. Biobehav. Rev. 2022, 136, 104596. [Google Scholar] [CrossRef]
- Barone, A.; Signoriello, S.; Latte, G.; Vellucci, L.; Giordano, G.; Avagliano, C.; Buonaguro, E.F.; Marmo, F.; Tomasetti, C.; Iasevoli, F.; et al. Modulation of glutamatergic functional connectivity by a prototypical antipsychotic: Translational inference from a postsynaptic density immediate-early gene-based network analysis. Behav. Brain Res. 2021, 404, 113160. [Google Scholar] [CrossRef]
- Hu, T.M.; Wang, Y.C.; Wu, C.L.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Multiple Rare Risk Coding Variants in Postsynaptic Density-Related Genes Associated With Schizophrenia Susceptibility. Front. Genet. 2020, 11, 524258. [Google Scholar] [CrossRef]
- Berdenis van Berlekom, A.; Muflihah, C.H.; Snijders, G.; MacGillavry, H.D.; Middeldorp, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr. Bull. 2020, 46, 374–386. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Avagliano, C.; Vellucci, L.; D’Ambrosio, L.; Manchia, M.; D’Urso, G.; Buonaguro, E.F.; Iasevoli, F. Translating preclinical findings in clinically relevant new antipsychotic targets: Focus on the glutamatergic postsynaptic density. Implications for treatment resistant schizophrenia. Neurosci. Biobehav. Rev. 2019, 107, 795–827. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 10, 89. [Google Scholar] [CrossRef]
- Bayés, À.; Collins, M.O.; Reig-Viader, R.; Gou, G.; Goulding, D.; Izquierdo, A.; Choudhary, J.S.; Emes, R.D.; Grant, S.G. Evolution of complexity in the zebrafish synapse proteome. Nat. Commun. 2017, 8, 14613. [Google Scholar] [CrossRef]
- Wilson, R.S.; Rauniyar, N.; Sakaue, F.; Lam, T.T.; Williams, K.R.; Nairn, A.C. Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD). Proteomes 2019, 7, 12. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Buonaguro, E.F.; Iasevoli, F. Serotonin-glutamate and serotonin-dopamine reciprocal interactions as putative molecular targets for novel antipsychotic treatments: From receptor heterodimers to postsynaptic scaffolding and effector proteins. Psychopharmacology 2013, 225, 1–19. [Google Scholar] [CrossRef]
- Lum, J.S.; Fernandez, F.; Matosin, N.; Andrews, J.L.; Huang, X.F.; Ooi, L.; Newell, K.A. Neurodevelopmental Expression Profile of Dimeric and Monomeric Group 1 mGluRs: Relevance to Schizophrenia Pathogenesis and Treatment. Sci. Rep. 2016, 6, 34391. [Google Scholar] [CrossRef]
- Suzuki, T.; Kametani, K.; Guo, W.; Li, W. Protein components of post-synaptic density lattice, a backbone structure for type I excitatory synapses. J. Neurochem. 2018, 144, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Beck, K.; Reis Marques, T.; Howes, O.D. Synaptic loss in schizophrenia: A meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 2019, 24, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Valtschanoff, J.G.; Weinberg, R.J. Laminar organization of the NMDA receptor complex within the postsynaptic density. J. Neurosci. 2001, 21, 1211–1217. [Google Scholar] [CrossRef]
- Boeckers, T.M. The postsynaptic density. Cell Tissue Res. 2006, 326, 409–422. [Google Scholar] [CrossRef]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfinger, E.D. ProSAP/Shank proteins–A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef]
- England, C.G.; Luo, H.; Cai, W. HaloTag technology: A versatile platform for biomedical applications. Bioconjugate Chem. 2015, 26, 975–986. [Google Scholar] [CrossRef]
- Kim, J.H.; Marton, J.; Ametamey, S.M.; Cumming, P. A Review of Molecular Imaging of Glutamate Receptors. Molecules 2020, 25, 4749. [Google Scholar] [CrossRef]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef]
- Syková, E.; Nicholson, C. Diffusion in brain extracellular space. Physiol. Rev. 2008, 88, 1277–1340. [Google Scholar] [CrossRef]
- Day, C.A.; Kenworthy, A.K. Tracking microdomain dynamics in cell membranes. Biochim. Biophys. Acta 2009, 1788, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Triller, A.; Choquet, D. New concepts in synaptic biology derived from single-molecule imaging. Neuron 2008, 59, 359–374. [Google Scholar] [CrossRef]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.; Pankratov, Y. ATP from synaptic terminals and astrocytes regulates NMDA receptors and synaptic plasticity through PSD-95 multi-protein complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef]
- Sheng, M.; Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Kojima, N.; Shirao, T. Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem. Int. 2007, 51, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Wegner, W.; Mott, A.C.; Grant, S.G.N.; Steffens, H.; Willig, K.I. In vivo STED microscopy visualizes PSD95 sub-structures and morphological changes over several hours in the mouse visual cortex. Sci. Rep. 2018, 8, 219. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Tang, C.; Verpelli, C.; Narayanan, R.; Stearns, M.H.; Xu, R.M.; Li, H.; Sala, C.; Hayashi, Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 2009, 137, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, E. The Shank family of scaffold proteins. J. Cell Sci. 2000, 113, 1851–1856. [Google Scholar] [CrossRef]
- Lim, S.; Naisbitt, S.; Yoon, J.; Hwang, J.I.; Suh, P.G.; Sheng, M.; Kim, E. Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 1999, 274, 29510–29518. [Google Scholar] [CrossRef] [Green Version]
- Zitzer, H.; Richter, D.; Kreienkamp, H.J. Agonist-dependent interaction of the rat somatostatin receptor subtype 2 with cortactin-binding protein 1. J. Biol. Chem. 1999, 274, 18153–18156. [Google Scholar] [CrossRef]
- Boeckers, T.M.; Kreutz, M.R.; Winter, C.; Zuschratter, W.; Smalla, K.H.; Sanmarti-Vila, L.; Wex, H.; Langnaese, K.; Bockmann, J.; Garner, C.C.; et al. Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J. Neurosci. 1999, 19, 6506–6518. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, H.J. Scaffolding proteins at the postsynaptic density: Shank as the architectural framework. Handb. Exp. Pharmacol. 2008, 15, 365–380. [Google Scholar] [CrossRef]
- Wang, X.; Bey, A.L.; Katz, B.M.; Badea, A.; Kim, N.; David, L.K.; Duffney, L.J.; Kumar, S.; Mague, S.D.; Hulbert, S.W.; et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 2016, 7, 11459. [Google Scholar] [CrossRef]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.J.; Reim, D.; Morello, N.; Orellana, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2017, 22, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.C.; Xiao, B.; Naisbitt, S.; Yuan, J.P.; Petralia, R.S.; Brakeman, P.; Doan, A.; Aakalu, V.K.; Lanahan, A.A.; Sheng, M.; et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 1999, 23, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Roussignol, G.; Ango, F.; Romorini, S.; Tu, J.C.; Sala, C.; Worley, P.F.; Bockaert, J.; Fagni, L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 2005, 25, 3560–3570. [Google Scholar] [CrossRef] [PubMed]
- Arons, M.H.; Thynne, C.J.; Grabrucker, A.M.; Li, D.; Schoen, M.; Cheyne, J.E.; Boeckers, T.M.; Montgomery, J.M.; Garner, C.C. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 2012, 32, 14966–14978. [Google Scholar] [CrossRef]
- Durand, C.M.; Perroy, J.; Loll, F.; Perrais, D.; Fagni, L.; Bourgeron, T.; Montcouquiol, M.; Sans, N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 2012, 17, 71–84. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Latte, G.; Tomasetti, C.; Iasevoli, F. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: Relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol. Neurobiol. 2014, 49, 484–511. [Google Scholar] [CrossRef]
- Guilmatre, A.; Huguet, G.; Delorme, R.; Bourgeron, T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 2014, 74, 113–122. [Google Scholar] [CrossRef]
- Grabrucker, A.M.; Schmeisser, M.J.; Schoen, M.; Boeckers, T.M. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 2011, 21, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Jutla, A.; Foss-Feig, J.; Veenstra-VanderWeele, J. Autism spectrum disorder and schizophrenia: An updated conceptual review. Autism Res. 2022, 15, 384–412. [Google Scholar] [CrossRef] [PubMed]
- Wöhr, M.; Roullet, F.I.; Hung, A.Y.; Sheng, M.; Crawley, J.N. Communication impairments in mice lacking Shank1: Reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS ONE 2011, 6, e20631. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef]
- Ha, S.; Lee, D.; Cho, Y.S.; Chung, C.; Yoo, Y.E.; Kim, J.; Lee, J.; Kim, W.; Kim, H.; Bae, Y.C.; et al. Cerebellar Shank2 Regulates Excitatory Synapse Density, Motor Coordination, and Specific Repetitive and Anxiety-Like Behaviors. J. Neurosci. 2016, 36, 12129–12143. [Google Scholar] [CrossRef]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.A.; Gao, X.; Fu, Z.; Feng, G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016, 530, 481–484. [Google Scholar] [CrossRef]
- Schmeisser, M.J.; Ey, E.; Wegener, S.; Bockmann, J.; Stempel, A.V.; Kuebler, A.; Janssen, A.L.; Udvardi, P.T.; Shiban, E.; Spilker, C.; et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012, 486, 256–260. [Google Scholar] [CrossRef]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabrucker, S.; Proepper, C.; Mangus, K.; Eckert, M.; Chhabra, R.; Schmeisser, M.J.; Boeckers, T.M.; Grabrucker, A.M. The PSD protein ProSAP2/Shank3 displays synapto-nuclear shuttling which is deregulated in a schizophrenia-associated mutation. Exp. Neurol. 2014, 253, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, M.J. Translational neurobiology in Shank mutant mice--model systems for neuropsychiatric disorders. Ann. Anat. Anat. Anz. 2015, 200, 115–117. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Champagne, N.; Lafrenière, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Côté, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef]
- Han, K.; Holder, J.L., Jr.; Schaaf, C.P.; Lu, H.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013, 503, 72–77. [Google Scholar] [CrossRef]
- Failla, P.; Romano, C.; Alberti, A.; Vasta, A.; Buono, S.; Castiglia, L.; Luciano, D.; Di Benedetto, D.; Fichera, M.; Galesi, O. Schizophrenia in a patient with subtelomeric duplication of chromosome 22q. Clin. Genet. 2007, 71, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Pang, K.; Kim, J.Y.; Ryu, J.R.; Kang, H.; Liu, Z.; Kim, W.K.; Sun, W.; Kim, H.; Han, K. Post-transcriptional regulation of SHANK3 expression by microRNAs related to multiple neuropsychiatric disorders. Mol. Brain 2015, 8, 74. [Google Scholar] [CrossRef]
- Lee, Y.; Zhang, Y.; Kim, S.; Han, K. Excitatory and inhibitory synaptic dysfunction in mania: An emerging hypothesis from animal model studies. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Föcking, M.; Lopez, L.M.; English, J.A.; Dicker, P.; Wolff, A.; Brindley, E.; Wynne, K.; Cagney, G.; Cotter, D.R. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol. Psychiatry 2015, 20, 424–432. [Google Scholar] [CrossRef]
- Balu, D.T.; Coyle, J.T. Chronic D-serine reverses arc expression and partially rescues dendritic abnormalities in a mouse model of NMDA receptor hypofunction. Neurochem. Int. 2014, 75, 76–78. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, H.J.; Luong, T.N.; Medina-Marino, A.; Schenker, L.; Khorosheva, E.; Indersmitten, T.; Gunapala, K.M.; Steele, A.D.; O’Dell, T.J.; Patterson, P.H.; et al. Deletion of densin-180 results in abnormal behaviors associated with mental illness and reduces mGluR5 and DISC1 in the postsynaptic density fraction. J. Neurosci. 2011, 31, 16194–16207. [Google Scholar] [CrossRef] [PubMed]
- Forrest, C.M.; Khalil, O.S.; Pisar, M.; Darlington, L.G.; Stone, T.W. Prenatal inhibition of the tryptophan-kynurenine pathway alters synaptic plasticity and protein expression in the rat hippocampus. Brain Res. 2013, 1504, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Hattori, T.; Nishida, T.; Hori, O.; Tohyama, M. Alterations in dendrite and spine morphology of cortical pyramidal neurons in DISC1-binding zinc finger protein (DBZ) knockout mice. Front. Neuroanat. 2015, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Su, Q.P.; Jin, D.; Yu, Y.; Huang, X.F. Prevention of Neurite Spine Loss Induced by Dopamine D2 Receptor Overactivation in Striatal Neurons. Front. Neurosci. 2020, 14, 642. [Google Scholar] [CrossRef]
- Cahill, M.E.; Jones, K.A.; Rafalovich, I.; Xie, Z.; Barros, C.S.; Müller, U.; Penzes, P. Control of interneuron dendritic growth through NRG1/erbB4-mediated kalirin-7 disinhibition. Mol. Psychiatry 2012, 17, 99–107. [Google Scholar] [CrossRef]
- Fujita-Jimbo, E.; Tanabe, Y.; Yu, Z.; Kojima, K.; Mori, M.; Li, H.; Iwamoto, S.; Yamagata, T.; Momoi, M.Y.; Momoi, T. The association of GPR85 with PSD-95-neuroligin complex and autism spectrum disorder: A molecular analysis. Mol. Autism 2015, 6, 17. [Google Scholar] [CrossRef]
- Li, R.; Chen, M.; Tian, H.; Li, G.; Wang, L.; Tu, W.; Chen, G.; Ping, J.; Zhuo, C.; Li, J. Association between ErbB4 gene function in synaptogenesis and schizophrenia pathogenesis. Biotechnol. Biotechnol. Equip. 2020, 34, 135–143. [Google Scholar] [CrossRef]
- Longart, M.; Chatani-Hinze, M.; Gonzalez, C.M.; Vullhorst, D.; Buonanno, A. Regulation of ErbB-4 endocytosis by neuregulin in GABAergic hippocampal interneurons. Brain Res. Bull. 2007, 73, 210–219. [Google Scholar] [CrossRef]
- Li, J.T.; Feng, Y.; Su, Y.A.; Wang, X.D.; Si, T.M. Enhanced interaction among ErbB4, PSD-95 and NMDAR by chronic MK-801 treatment is associated with behavioral abnormalities. Pharmacol. Biochem. Behav. 2013, 108, 44–53. [Google Scholar] [CrossRef]
- Ting, A.K.; Chen, Y.; Wen, L.; Yin, D.M.; Shen, C.; Tao, Y.; Liu, X.; Xiong, W.C.; Mei, L. Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons. J. Neurosci. 2011, 31, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Balu, D.T.; Coyle, J.T. Glutamate receptor composition of the post-synaptic density is altered in genetic mouse models of NMDA receptor hypo- and hyperfunction. Brain Res. 2011, 1392, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Wang, H.Y.; Borgmann-Winter, K.E.; MacDonald, M.L.; Kaprielian, H.; Stucky, A.; Kvasic, J.; Egbujo, C.; Ray, R.; Talbot, K.; et al. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol. Psychiatry 2015, 20, 1091–1100. [Google Scholar] [CrossRef]
- Coba, M.P.; Ramaker, M.J.; Ho, E.V.; Thompson, S.L.; Komiyama, N.H.; Grant, S.G.N.; Knowles, J.A.; Dulawa, S.C. Dlgap1 knockout mice exhibit alterations of the postsynaptic density and selective reductions in sociability. Sci. Rep. 2018, 8, 2281. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.A.; Gao, W.J. PSD-95 deficiency disrupts PFC-associated function and behavior during neurodevelopment. Sci. Rep. 2019, 9, 9486. [Google Scholar] [CrossRef]
- Culotta, L.; Scalmani, P.; Vinci, E.; Terragni, B.; Sessa, A.; Broccoli, V.; Mantegazza, M.; Boeckers, T.; Verpelli, C. SULT4A1 Modulates Synaptic Development and Function by Promoting the Formation of PSD-95/NMDAR Complex. J. Neurosci. 2020, 40, 7013–7026. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Collins, M.O.; Uren, R.T.; Kopanitsa, M.V.; Komiyama, N.H.; Croning, M.D.; Zografos, L.; Armstrong, J.D.; Choudhary, J.S.; Grant, S.G. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol. 2009, 5, 269. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Morini, R.; Corradini, I.; Antonucci, F.; Trepte, P.; Edry, E.; Sharma, V.; Papale, A.; Pozzi, D.; Defilippi, P.; et al. Reduced SNAP-25 increases PSD-95 mobility and impairs spine morphogenesis. Cell Death Differ. 2015, 22, 1425–1436. [Google Scholar] [CrossRef]
- Ganguly, P.; Holland, F.H.; Brenhouse, H.C. Functional Uncoupling NMDAR NR2A Subunit from PSD-95 in the Prefrontal Cortex: Effects on Behavioral Dysfunction and Parvalbumin Loss after Early-Life Stress. Neuropsychopharmacology 2015, 40, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef]
- Jin, C.; Kang, H.; Ryu, J.R.; Kim, S.; Zhang, Y.; Lee, Y.; Kim, Y.; Han, K. Integrative Brain Transcriptome Analysis Reveals Region-Specific and Broad Molecular Changes in Shank3-Overexpressing Mice. Front. Mol. Neurosci. 2018, 11, 250. [Google Scholar] [CrossRef] [Green Version]
- Loomis, C.; Stephens, A.; Janicot, R.; Baqai, U.; Drebushenko, L.; Round, J. Identification of MAGUK scaffold proteins as intracellular binding partners of synaptic adhesion protein Slitrk2. Mol. Cell. Neurosci. 2020, 103, 103465. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.M.; Paul, B.D.; Fu, C.; Hu, S.; Zhu, H.; Blackshaw, S.; Wolosker, H.; Snyder, S.H. Serine racemase regulated by binding to stargazin and PSD-95: Potential N-methyl-D-aspartate-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (NMDA-AMPA) glutamate neurotransmission cross-talk. J. Biol. Chem. 2014, 289, 29631–29641. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Torres, N.I.; Vázquez-Hernández, N.; Martín-Amaya-Barajas, F.L.; Flores-Soto, M.; González-Burgos, I. Ibotenic acid induced lesions impair the modulation of dendritic spine plasticity in the prefrontal cortex and amygdala, a phenomenon that underlies working memory and social behavior. Eur. J. Pharmacol. 2021, 896, 173883. [Google Scholar] [CrossRef] [PubMed]
- McEachern, E.P.; Coley, A.A.; Yang, S.S.; Gao, W.J. PSD-95 deficiency alters GABAergic inhibition in the prefrontal cortex. Neuropharmacology 2020, 179, 108277. [Google Scholar] [CrossRef]
- Oyagi, A.; Oida, Y.; Kakefuda, K.; Shimazawa, M.; Shioda, N.; Moriguchi, S.; Kitaichi, K.; Nanba, D.; Yamaguchi, K.; Furuta, Y.; et al. Generation and characterization of conditional heparin-binding EGF-like growth factor knockout mice. PLoS ONE 2009, 4, e7461. [Google Scholar] [CrossRef]
- Dunn, H.A.; Walther, C.; Godin, C.M.; Hall, R.A.; Ferguson, S.S. Role of SAP97 protein in the regulation of corticotropin-releasing factor receptor 1 endocytosis and extracellular signal-regulated kinase 1/2 signaling. J. Biol. Chem. 2013, 288, 15023–15034. [Google Scholar] [CrossRef]
- Gupta, P.; Uner, O.E.; Nayak, S.; Grant, G.R.; Kalb, R.G. SAP97 regulates behavior and expression of schizophrenia risk enriched gene sets in mouse hippocampus. PLoS ONE 2018, 13, e0200477. [Google Scholar] [CrossRef]
- Ishida, H.; Skorobogatov, A.; Yamniuk, A.P.; Vogel, H.J. Solution structures of the SH3 domains from Shank scaffold proteins and their interactions with Cav1.3 calcium channels. FEBS Lett. 2018, 592, 2786–2797. [Google Scholar] [CrossRef]
- Li, J.; Wilkinson, B.; Clementel, V.A.; Hou, J.; O’Dell, T.J.; Coba, M.P. Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci. Signal. 2016, 9, 8. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W.; Yang, H.; Howrigan, D.P.; Wilkinson, B.; Souaiaia, T.; Evgrafov, O.V.; Genovese, G.; Clementel, V.A.; Tudor, J.C.; et al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat. Neurosci. 2017, 20, 1150–1161. [Google Scholar] [CrossRef]
- Ponna, S.K.; Ruskamo, S.; Myllykoski, M.; Keller, C.; Boeckers, T.M.; Kursula, P. Structural basis for PDZ domain interactions in the post-synaptic density scaffolding protein Shank3. J. Neurochem. 2018, 145, 449–463. [Google Scholar] [CrossRef]
- Critchlow, H.M.; Maycox, P.R.; Skepper, J.N.; Krylova, O. Clozapine and haloperidol differentially regulate dendritic spine formation and synaptogenesis in rat hippocampal neurons. Mol. Cell. Neurosci. 2006, 32, 356–365. [Google Scholar] [CrossRef]
- Leucht, S.; Corves, C.; Arbter, D.; Engel, R.R.; Li, C.; Davis, J.M. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet 2009, 373, 31–41. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Tomasetti, C.; Cicale, M.; Yuan, P.X.; Manji, H.K. Chronic treatment with lithium or valproate modulates the expression of Homer1b/c and its related genes Shank and Inositol 1,4,5-trisphosphate receptor. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2012, 22, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Roussignol, G.; Meldolesi, J.; Fagni, L. Key role of the postsynaptic density scaffold proteins Shank and Homer in the functional architecture of Ca2+ homeostasis at dendritic spines in hippocampal neurons. J. Neurosci. 2005, 25, 4587–4592. [Google Scholar] [CrossRef]
- Buonaguro, E.F.; Tomasetti, C.; Chiodini, P.; Marmo, F.; Latte, G.; Rossi, R.; Avvisati, L.; Iasevoli, F.; de Bartolomeis, A. Postsynaptic density protein transcripts are differentially modulated by minocycline alone or in add-on to haloperidol: Implications for treatment resistant schizophrenia. J. Psychopharmacol. 2017, 31, 406–417. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Iasevoli, F.; Marmo, F.; Buonaguro, E.F.; Eramo, A.; Rossi, R.; Avvisati, L.; Latte, G.; Tomasetti, C. Progressive recruitment of cortical and striatal regions by inducible postsynaptic density transcripts after increasing doses of antipsychotics with different receptor profiles: Insights for psychosis treatment. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2015, 25, 566–582. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Iasevoli, F.; Marmo, F.; Buonaguro, E.F.; Avvisati, L.; Latte, G.; Tomasetti, C. Nicotine and caffeine modulate haloperidol-induced changes in postsynaptic density transcripts expression: Translational insights in psychosis therapy and treatment resistance. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2018, 28, 538–559. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; Marmo, F.; Bravi, D.; Arnt, J.; de Bartolomeis, A. Divergent acute and chronic modulation of glutamatergic postsynaptic density genes expression by the antipsychotics haloperidol and sertindole. Psychopharmacology 2010, 212, 329–344. [Google Scholar] [CrossRef]
- Kirkpatrick, B.; Xu, L.; Cascella, N.; Ozeki, Y.; Sawa, A.; Roberts, R.C. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J. Comp. Neurol. 2006, 497, 436–450. [Google Scholar] [CrossRef]
- Matosin, N.; Fernandez-Enright, F.; Lum, J.S.; Engel, M.; Andrews, J.L.; Gassen, N.C.; Wagner, K.V.; Schmidt, M.V.; Newell, K.A. Molecular evidence of synaptic pathology in the CA1 region in schizophrenia. NPJ Schizophr. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, E.F.; Iasevoli, F.; Marmo, F.; Eramo, A.; Latte, G.; Avagliano, C.; Tomasetti, C.; de Bartolomeis, A. Re-arrangements of gene transcripts at glutamatergic synapses after prolonged treatments with antipsychotics: A putative link with synaptic remodeling. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2017, 76, 29–41. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Aloj, L.; Ambesi-Impiombato, A.; Bravi, D.; Caracò, C.; Muscettola, G.; Barone, P. Acute administration of antipsychotics modulates Homer striatal gene expression differentially. Brain Res. Mol. Brain Res. 2002, 98, 124–129. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Sarappa, C.; Buonaguro, E.F.; Marmo, F.; Eramo, A.; Tomasetti, C.; Iasevoli, F. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: Role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2013, 46, 1–12. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Errico, F.; Aceto, G.; Tomasetti, C.; Usiello, A.; Iasevoli, F. D-aspartate dysregulation in Ddo(-/-) mice modulates phencyclidine-induced gene expression changes of postsynaptic density molecules in cortex and striatum. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2015, 62, 35–43. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Marmo, F.; Buonaguro, E.F.; Latte, G.; Tomasetti, C.; Iasevoli, F. Switching antipsychotics: Imaging the differential effect on the topography of postsynaptic density transcripts in antipsychotic-naïve vs. antipsychotic-exposed rats. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2016, 70, 24–38. [Google Scholar] [CrossRef]
- Iasevoli, F.; Polese, D.; Ambesi-Impiombato, A.; Muscettola, G.; de Bartolomeis, A. Ketamine-related expression of glutamatergic postsynaptic density genes: Possible implications in psychosis. Neurosci. Lett. 2007, 416, 1–5. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; Ambesi-Impiombato, A.; Muscettola, G.; de Bartolomeis, A. Dopamine receptor subtypes contribution to Homer1a induction: Insights into antipsychotic molecular action. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2009, 33, 813–821. [Google Scholar] [CrossRef]
- Iasevoli, F.; Ambesi-Impiombato, A.; Fiore, G.; Panariello, F.; Muscettola, G.; de Bartolomeis, A. Pattern of acute induction of Homer1a gene is preserved after chronic treatment with first- and second-generation antipsychotics: Effect of short-term drug discontinuation and comparison with Homer1a-interacting genes. J. Psychopharmacol. 2011, 25, 875–887. [Google Scholar] [CrossRef]
- Iasevoli, F.; Buonaguro, E.F.; Sarappa, C.; Marmo, F.; Latte, G.; Rossi, R.; Eramo, A.; Tomasetti, C.; de Bartolomeis, A. Regulation of postsynaptic plasticity genes’ expression and topography by sustained dopamine perturbation and modulation by acute memantine: Relevance to schizophrenia. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2014, 54, 299–314. [Google Scholar] [CrossRef]
- Iasevoli, F.; Buonaguro, E.F.; Avagliano, C.; Barone, A.; Eramo, A.; Vellucci, L.; de Bartolomeis, A. The Effects of Antipsychotics on the Synaptic Plasticity Gene Homer1a Depend on a Combination of Their Receptor Profile, Dose, Duration of Treatment, and Brain Regions Targeted. Int. J. Mol. Sci. 2020, 21, 5555. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.G.; Wang, H.Y.; Cho, D.S.; Talbot, K.; Gur, R.E.; Berrettini, W.H.; Bakshi, K.; Kamins, J.; Borgmann-Winter, K.E.; Siegel, S.J.; et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 2006, 12, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Morrison, P.D.; Pilowsky, L.S. Schizophrenia: More evidence for less glutamate. Expert Rev. Neurother. 2007, 7, 29–31. [Google Scholar] [CrossRef] [PubMed]
- du Bois, T.M.; Newell, K.A.; Huang, X.F. Perinatal phencyclidine treatment alters neuregulin 1/erbB4 expression and activation in later life. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2012, 22, 356–363. [Google Scholar] [CrossRef]
- Balan, S.; Yamada, K.; Hattori, E.; Iwayama, Y.; Toyota, T.; Ohnishi, T.; Maekawa, M.; Toyoshima, M.; Iwata, Y.; Suzuki, K.; et al. Population-specific haplotype association of the postsynaptic density gene DLG4 with schizophrenia, in family-based association studies. PLoS ONE 2013, 8, e70302. [Google Scholar] [CrossRef]
- Catts, V.S.; Derminio, D.S.; Hahn, C.G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. NPJ Schizophr. 2015, 1, 15037. [Google Scholar] [CrossRef]
- Funk, A.J.; McCullumsmith, R.E.; Haroutunian, V.; Meador-Woodruff, J.H. Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology 2012, 37, 896–905. [Google Scholar] [CrossRef]
- Funk, A.J.; Mielnik, C.A.; Koene, R.; Newburn, E.; Ramsey, A.J.; Lipska, B.K.; McCullumsmith, R.E. Postsynaptic Density-95 Isoform Abnormalities in Schizophrenia. Schizophr. Bull. 2017, 43, 891–899. [Google Scholar] [CrossRef]
- Kawashima, R.; Ohnuma, T.; Shibata, N.; Arai, H. No genetic association between postsynaptic density-95 gene polymorphisms and schizophrenia. Neurosci. Lett. 2006, 400, 168–171. [Google Scholar] [CrossRef]
- Tsai, S.J.; Hong, C.J.; Cheng, C.Y.; Liao, D.L.; Liou, Y.J. Association study of polymorphisms in post-synaptic density protein 95 (PSD-95) with schizophrenia. J. Neural Transm. 2007, 114, 423–426. [Google Scholar] [CrossRef]
- Xing, J.; Kimura, H.; Wang, C.; Ishizuka, K.; Kushima, I.; Arioka, Y.; Yoshimi, A.; Nakamura, Y.; Shiino, T.; Oya-Ito, T.; et al. Resequencing and Association Analysis of Six PSD-95-Related Genes as Possible Susceptibility Genes for Schizophrenia and Autism Spectrum Disorders. Sci. Rep. 2016, 6, 27491. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, L.V.; Bakir, B.; Haroutunian, V.; Meador-Woodruff, J.H. Expression of the NR2B-NMDA receptor trafficking complex in prefrontal cortex from a group of elderly patients with schizophrenia. Schizophr. Res. 2010, 119, 198–209. [Google Scholar] [CrossRef]
- Kristiansen, L.V.; Beneyto, M.; Haroutunian, V.; Meador-Woodruff, J.H. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 2006, 11, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Matas, E.; John Francis William, D.; Toro, C.T. Abnormal expression of post-synaptic proteins in prefrontal cortex of patients with schizophrenia. Neurosci. Lett. 2021, 745, 135629. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Louneva, N.; Cohen, J.W.; Kazi, H.; Blake, D.J.; Arnold, S.E. Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS ONE 2011, 6, e16886. [Google Scholar] [CrossRef]
- Toro, C.; Deakin, J.F. NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr. Res. 2005, 80, 323–330. [Google Scholar] [CrossRef]
- Lin, H.; Jacobi, A.A.; Anderson, S.A.; Lynch, D.R. D-Serine and Serine Racemase Are Associated with PSD-95 and Glutamatergic Synapse Stability. Front. Cell. Neurosci. 2016, 10, 34. [Google Scholar] [CrossRef]
- Reumann, R.; Vierk, R.; Zhou, L.; Gries, F.; Kraus, V.; Mienert, J.; Romswinkel, E.; Morellini, F.; Ferrer, I.; Nicolini, C.; et al. The serine protease inhibitor neuroserpin is required for normal synaptic plasticity and regulates learning and social behavior. Learn. Mem. 2017, 24, 650–659. [Google Scholar] [CrossRef]
- Takaki, M.; Kodama, M.; Mizuki, Y.; Kawai, H.; Yoshimura, B.; Kishimoto, M.; Sakamoto, S.; Okahisa, Y.; Yamada, N. Effects of the antipsychotics haloperidol, clozapine, and aripiprazole on the dendritic spine. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2018, 28, 610–619. [Google Scholar] [CrossRef]
- Xu, Y.; Deng, C.; Zheng, Y.; Liu, N.; Fu, B. Applying vinpocetine to reverse synaptic ultrastructure by regulating BDNF-related PSD-95 in alleviating schizophrenia-like deficits in rat. Compr. Psychiatry 2019, 94, 152122. [Google Scholar] [CrossRef]
- Li, J.M.; Lu, C.L.; Cheng, M.C.; Luu, S.U.; Hsu, S.H.; Hu, T.M.; Tsai, H.Y.; Chen, C.H. Role of the DLGAP2 gene encoding the SAP90/PSD-95-associated protein 2 in schizophrenia. PLoS ONE 2014, 9, e85373. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, K.; Iritani, S.; Makifuchi, T.; Shirakawa, O.; Kitamura, N.; Maeda, K.; Nakamura, R.; Niizato, K.; Watanabe, M.; Kakita, A.; et al. Selective reduction of a PDZ protein, SAP-97, in the prefrontal cortex of patients with chronic schizophrenia. J. Neurochem. 2002, 83, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Beneyto, M.; Meador-Woodruff, J.H. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 2008, 33, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Clinton, S.M.; Haroutunian, V.; Meador-Woodruff, J.H. Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J. Neurochem. 2006, 98, 1114–1125. [Google Scholar] [CrossRef]
- Kristiansen, L.V.; Meador-Woodruff, J.H. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr. Res. 2005, 78, 87–93. [Google Scholar] [CrossRef]
- McCullumsmith, R.E.; Kristiansen, L.V.; Beneyto, M.; Scarr, E.; Dean, B.; Meador-Woodruff, J.H. Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res. 2007, 1127, 108–118. [Google Scholar] [CrossRef]
- Mueller, H.T.; Haroutunian, V.; Davis, K.L.; Meador-Woodruff, J.H. Expression of the ionotropic glutamate receptor subunits and NMDA receptor-associated intracellular proteins in the substantia nigra in schizophrenia. Brain Res. Mol. Brain Res. 2004, 121, 60–69. [Google Scholar] [CrossRef]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef]
- Brocos-Mosquera, I.; Gabilondo, A.M.; Meana, J.J.; Callado, L.F.; Erdozain, A.M. Spinophilin expression in postmortem prefrontal cortex of schizophrenic subjects: Effects of antipsychotic treatment. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2021, 42, 12–21. [Google Scholar] [CrossRef]
- Catts, V.S.; Weickert, C.S. Gene expression analysis implicates a death receptor pathway in schizophrenia pathology. PLoS ONE 2012, 7, e35511. [Google Scholar] [CrossRef]
- Cheng, D.; Hoogenraad, C.C.; Rush, J.; Ramm, E.; Schlager, M.A.; Duong, D.M.; Xu, P.; Wijayawardana, S.R.; Hanfelt, J.; Nakagawa, T.; et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteom. 2006, 5, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.O.; Hunt, C.A.; Kennedy, M.B. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 1992, 9, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chetkovich, D.M.; Petralia, R.S.; Sweeney, N.T.; Kawasaki, Y.; Wenthold, R.J.; Bredt, D.S.; Nicoll, R.A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Kornau, H.C.; Schenker, L.T.; Kennedy, M.B.; Seeburg, P.H. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 1995, 269, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef]
- Zhang, H.; Etherington, L.A.; Hafner, A.S.; Belelli, D.; Coussen, F.; Delagrange, P.; Chaouloff, F.; Spedding, M.; Lambert, J.J.; Choquet, D.; et al. Regulation of AMPA receptor surface trafficking and synaptic plasticity by a cognitive enhancer and antidepressant molecule. Mol. Psychiatry 2013, 18, 471–484. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Irie, M.; Hata, Y.; Takeuchi, M.; Ichtchenko, K.; Toyoda, A.; Hirao, K.; Takai, Y.; Rosahl, T.W.; Südhof, T.C. Binding of neuroligins to PSD-95. Science 1997, 277, 1511–1515. [Google Scholar] [CrossRef]
- Kim, E.; Naisbitt, S.; Hsueh, Y.P.; Rao, A.; Rothschild, A.; Craig, A.M.; Sheng, M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J. Cell Biol. 1997, 136, 669–678. [Google Scholar] [CrossRef]
- Kim, E.; Niethammer, M.; Rothschild, A.; Jan, Y.N.; Sheng, M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 1995, 378, 85–88. [Google Scholar] [CrossRef]
- Glantz, L.A.; Gilmore, J.H.; Hamer, R.M.; Lieberman, J.A.; Jarskog, L.F. Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience 2007, 149, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Craven, S.E.; El-Husseini, A.E.; Bredt, D.S. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron 1999, 22, 497–509. [Google Scholar] [CrossRef] [PubMed]
- El-Husseini, A.E.; Craven, S.E.; Chetkovich, D.M.; Firestein, B.L.; Schnell, E.; Aoki, C.; Bredt, D.S. Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J. Cell Biol. 2000, 148, 159–172. [Google Scholar] [CrossRef]
- Fukata, Y.; Dimitrov, A.; Boncompain, G.; Vielemeyer, O.; Perez, F.; Fukata, M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J. Cell Biol. 2013, 202, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Topinka, J.R.; Bredt, D.S. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 1998, 20, 125–134. [Google Scholar] [CrossRef]
- Funke, L.; Dakoji, S.; Bredt, D.S. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu. Rev. Biochem. 2005, 74, 219–245. [Google Scholar] [CrossRef]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef]
- Gray, N.W.; Weimer, R.M.; Bureau, I.; Svoboda, K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006, 4, e370. [Google Scholar] [CrossRef]
- Dumas, T.C. Developmental regulation of cognitive abilities: Modified composition of a molecular switch turns on associative learning. Prog. Neurobiol. 2005, 76, 189–211. [Google Scholar] [CrossRef]
- Monaco, S.A.; Gulchina, Y.; Gao, W.J. NR2B subunit in the prefrontal cortex: A double-edged sword for working memory function and psychiatric disorders. Neurosci. Biobehav. Rev. 2015, 56, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Coley, A.A.; Gao, W.J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef]
- Disciglio, V.; Lo Rizzo, C.; Mencarelli, M.A.; Mucciolo, M.; Marozza, A.; Di Marco, C.; Massarelli, A.; Canocchi, V.; Baldassarri, M.; Ndoni, E.; et al. Interstitial 22q13 deletions not involving SHANK3 gene: A new contiguous gene syndrome. Am. J. Med. Genet. A 2014, 164a, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Mitz, A.R.; Philyaw, T.J.; Boccuto, L.; Shcheglovitov, A.; Sarasua, S.M.; Kaufmann, W.E.; Thurm, A. Identification of 22q13 genes most likely to contribute to Phelan McDermid syndrome. Eur. J. Hum. Genet. 2018, 26, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Ziats, C.A.; Grosvenor, L.P.; Sarasua, S.M.; Thurm, A.E.; Swedo, S.E.; Mahfouz, A.; Rennert, O.M.; Ziats, M.N. Functional genomics analysis of Phelan-McDermid syndrome 22q13 region during human neurodevelopment. PLoS ONE 2019, 14, e0213921. [Google Scholar] [CrossRef]
- Idris, M.; Butcher, N.J.; Minchin, R.F. The MBNL/CELF Splicing Factors Regulate Cytosolic Sulfotransferase 4A1 Protein Expression during Cell Differentiation. Drug Metab. Dispos. Biol. Fate Chem. 2019, 47, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Hossain, M.I.; Andrabi, S.A.; Falany, C.N. Generation and Characterization of SULT4A1 Mutant Mouse Models. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 41–45. [Google Scholar] [CrossRef]
- Futamura, T.; Toyooka, K.; Iritani, S.; Niizato, K.; Nakamura, R.; Tsuchiya, K.; Someya, T.; Kakita, A.; Takahashi, H.; Nawa, H. Abnormal expression of epidermal growth factor and its receptor in the forebrain and serum of schizophrenic patients. Mol. Psychiatry 2002, 7, 673–682. [Google Scholar] [CrossRef]
- Abbas, A.I.; Yadav, P.N.; Yao, W.D.; Arbuckle, M.I.; Grant, S.G.; Caron, M.G.; Roth, B.L. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J. Neurosci. 2009, 29, 7124–7136. [Google Scholar] [CrossRef]
- St Clair, D.; Blackwood, D.; Muir, W.; Carothers, A.; Walker, M.; Spowart, G.; Gosden, C.; Evans, H.J. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990, 336, 13–16. [Google Scholar] [CrossRef]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef]
- Horner, A.E.; Norris, R.H.; McLaren-Jones, R.; Alexander, L.; Komiyama, N.H.; Grant, S.G.N.; Nithianantharajah, J.; Kopanitsa, M.V. Learning and reaction times in mouse touchscreen tests are differentially impacted by mutations in genes encoding postsynaptic interacting proteins SYNGAP1, NLGN3, DLGAP1, DLGAP2 and SHANK2. Genes Brain Behav. 2021, 20, e12723. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Kato, H.; Arai, H.; Faull, R.L.; McKenna, P.J.; Emson, P.C. Gene expression of PSD95 in prefrontal cortex and hippocampus in schizophrenia. Neuroreport 2000, 11, 3133–3137. [Google Scholar] [CrossRef] [PubMed]
- Feyder, M.; Karlsson, R.M.; Mathur, P.; Lyman, M.; Bock, R.; Momenan, R.; Munasinghe, J.; Scattoni, M.L.; Ihne, J.; Camp, M.; et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am. J. Psychiatry 2010, 167, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef]
- Hodgkinson, C.A.; Goldman, D.; Jaeger, J.; Persaud, S.; Kane, J.M.; Lipsky, R.H.; Malhotra, A.K. Disrupted in schizophrenia 1 (DISC1): Association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am. J. Hum. Genet. 2004, 75, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.C.; Carlyle, B.C.; Bradshaw, N.J.; Porteous, D.J. DISC1: Structure, Function, and Therapeutic Potential for Major Mental Illness. ACS Chem. Neurosci. 2011, 2, 609–632. [Google Scholar] [CrossRef]
- Taylor, M.S.; Devon, R.S.; Millar, J.K.; Porteous, D.J. Evolutionary constraints on the Disrupted in Schizophrenia locus. Genomics 2003, 81, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Duff, B.J.; Macritchie, K.A.N.; Moorhead, T.W.J.; Lawrie, S.M.; Blackwood, D.H.R. Human brain imaging studies of DISC1 in schizophrenia, bipolar disorder and depression: A systematic review. Schizophr. Res. 2013, 147, 1–13. [Google Scholar] [CrossRef]
- Callicott, J.H.; Straub, R.E.; Pezawas, L.; Egan, M.F.; Mattay, V.S.; Hariri, A.R.; Verchinski, B.A.; Meyer-Lindenberg, A.; Balkissoon, R.; Kolachana, B.; et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 8627–8632. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Scannavini, M.; Falasca, A.; Hakim, G.; Busi, C.; Mattioli, A.; Fiume, L. Reduction of simian virus 40 growth by a mitotic inhibitor extracted from liver. Evidence that the inhibitor is arginase. Annali Sclavo 1981, 23, 162–168. [Google Scholar] [PubMed]
- Ishizuka, K.; Paek, M.; Kamiya, A.; Sawa, A. A review of Disrupted-In-Schizophrenia-1 (DISC1): Neurodevelopment, cognition, and mental conditions. Biol. Psychiatry 2006, 59, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Chubb, J.E.; Bradshaw, N.J.; Soares, D.C.; Porteous, D.J.; Millar, J.K. The DISC locus in psychiatric illness. Mol. Psychiatry 2008, 13, 36–64. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.F.; Bartos, M. Disrupted-in-schizophrenia-1 is required for normal pyramidal cell-interneuron communication and assembly dynamics in the prefrontal cortex. eLife 2022, 11, e79471. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Baskaran, R.; Tsao, C.Y.; Tuan, L.H.; Siow, P.F.; Palani, M.; Lee, L.J.; Liu, C.M.; Hwu, H.G.; Lee, L.J. Chronic N-Acetylcysteine Treatment Prevents Amphetamine-Induced Hyperactivity in Heterozygous Disc1 Mutant Mice, a Putative Prodromal Schizophrenia Animal Model. Int. J. Mol. Sci. 2022, 23, 9419. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Asai, N.; Namba, T.; Wang, Y.; Kato, T.; Tanaka, M.; Tatsumi, H.; Taya, S.; Tsuboi, D.; Kuroda, K.; et al. Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron 2009, 63, 774–787. [Google Scholar] [CrossRef]
- Shinoda, T.; Taya, S.; Tsuboi, D.; Hikita, T.; Matsuzawa, R.; Kuroda, S.; Iwamatsu, A.; Kaibuchi, K. DISC1 regulates neurotrophin-induced axon elongation via interaction with Grb2. J. Neurosci. 2007, 27, 4–14. [Google Scholar] [CrossRef]
- Kamiya, A.; Kubo, K.; Tomoda, T.; Takaki, M.; Youn, R.; Ozeki, Y.; Sawamura, N.; Park, U.; Kudo, C.; Okawa, M.; et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 2005, 7, 1167–1178. [Google Scholar] [CrossRef]
- Miyoshi, K.; Honda, A.; Baba, K.; Taniguchi, M.; Oono, K.; Fujita, T.; Kuroda, S.; Katayama, T.; Tohyama, M. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol. Psychiatry 2003, 8, 685–694. [Google Scholar] [CrossRef]
- Taya, S.; Shinoda, T.; Tsuboi, D.; Asaki, J.; Nagai, K.; Hikita, T.; Kuroda, S.; Kuroda, K.; Shimizu, M.; Hirotsune, S.; et al. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 2007, 27, 15–26. [Google Scholar] [CrossRef]
- Morris, J.A.; Kandpal, G.; Ma, L.; Austin, C.P. DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: Regulation and loss of interaction with mutation. Hum. Mol. Genet. 2003, 12, 1591–1608. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Tan, P.L.; Kubo, K.; Engelhard, C.; Ishizuka, K.; Kubo, A.; Tsukita, S.; Pulver, A.E.; Nakajima, K.; Cascella, N.G.; et al. Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: A candidate for psychiatric illnesses. Arch. Gen. Psychiatry 2008, 65, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Hashimoto, R.; Ohi, K.; Yamaguti, K.; Nakatomi, Y.; Yasuda, Y.; Kamino, K.; Takeda, M.; Tajima, S.; Kuratsune, H.; et al. A functional polymorphism in the disrupted-in schizophrenia 1 gene is associated with chronic fatigue syndrome. Life Sci. 2010, 86, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Katayama, T.; Tohyama, M.; Ogawa, N. DISC1 localizes to the centrosome by binding to kendrin. Biochem. Biophys. Res. Commun. 2004, 317, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Matsuzaki, S.; Hattori, T.; Kumamoto, N.; Miyoshi, K.; Katayama, T.; Tohyama, M. DISC1-kendrin interaction is involved in centrosomal microtubule network formation. Biochem. Biophys. Res. Commun. 2008, 377, 1051–1056. [Google Scholar] [CrossRef]
- Kamiya, A.; Tomoda, T.; Chang, J.; Takaki, M.; Zhan, C.; Morita, M.; Cascio, M.B.; Elashvili, S.; Koizumi, H.; Takanezawa, Y.; et al. DISC1-NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum. Mol. Genet. 2006, 15, 3313–3323. [Google Scholar] [CrossRef]
- Bradshaw, N.J.; Ogawa, F.; Antolin-Fontes, B.; Chubb, J.E.; Carlyle, B.C.; Christie, S.; Claessens, A.; Porteous, D.J.; Millar, J.K. DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem. Biophys. Res. Commun. 2008, 377, 1091–1096. [Google Scholar] [CrossRef]
- Marley, A.; von Zastrow, M. DISC1 regulates primary cilia that display specific dopamine receptors. PLoS ONE 2010, 5, e10902. [Google Scholar] [CrossRef]
- Brandon, N.J.; Schurov, I.; Camargo, L.M.; Handford, E.J.; Duran-Jimeniz, B.; Hunt, P.; Millar, J.K.; Porteous, D.J.; Shearman, M.S.; Whiting, P.J. Subcellular targeting of DISC1 is dependent on a domain independent from the Nudel binding site. Mol. Cell. Neurosci. 2005, 28, 613–624. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Suth, S.; Luth, E.S.; Sawa, A.; Selkoe, D.J. Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J. Neurosci. 2010, 30, 10431–10440. [Google Scholar] [CrossRef] [Green Version]
- Ozeki, Y.; Tomoda, T.; Kleiderlein, J.; Kamiya, A.; Bord, L.; Fujii, K.; Okawa, M.; Yamada, N.; Hatten, M.E.; Snyder, S.H.; et al. Disrupted-in-Schizophrenia-1 (DISC-1): Mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc. Natl. Acad. Sci. USA 2003, 100, 289–294. [Google Scholar] [CrossRef] [PubMed]
- James, R.; Adams, R.R.; Christie, S.; Buchanan, S.R.; Porteous, D.J.; Millar, J.K. Disrupted in Schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol. Cell. Neurosci. 2004, 26, 112–122. [Google Scholar] [CrossRef]
- Park, Y.U.; Jeong, J.; Lee, H.; Mun, J.Y.; Kim, J.H.; Lee, J.S.; Nguyen, M.D.; Han, S.S.; Suh, P.G.; Park, S.K. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc. Natl. Acad. Sci. USA 2010, 107, 17785–17790. [Google Scholar] [CrossRef] [PubMed]
- Atkin, T.A.; MacAskill, A.F.; Brandon, N.J.; Kittler, J.T. Disrupted in Schizophrenia-1 regulates intracellular trafficking of mitochondria in neurons. Mol. Psychiatry 2011, 16, 122–124. [Google Scholar] [CrossRef]
- Millar, J.K.; James, R.; Christie, S.; Porteous, D.J. Disrupted in schizophrenia 1 (DISC1): Subcellular targeting and induction of ring mitochondria. Mol. Cell. Neurosci. 2005, 30, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.; Evgrafov, O.V.; Zheng, D.; Hartel, N.; Knowles, J.A.; Graham, N.A.; Ichida, J.K.; Coba, M.P. Endogenous Cell Type-Specific Disrupted in Schizophrenia 1 Interactomes Reveal Protein Networks Associated With Neurodevelopmental Disorders. Biol. Psychiatry 2019, 85, 305–316. [Google Scholar] [CrossRef]
- Camargo, L.M.; Collura, V.; Rain, J.C.; Mizuguchi, K.; Hermjakob, H.; Kerrien, S.; Bonnert, T.P.; Whiting, P.J.; Brandon, N.J. Disrupted in Schizophrenia 1 Interactome: Evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry 2007, 12, 74–86. [Google Scholar] [CrossRef]
- Zamarbide, M.; Mossa, A.; Muñoz-Llancao, P.; Wilkinson, M.K.; Pond, H.L.; Oaks, A.W.; Manzini, M.C. Male-Specific cAMP Signaling in the Hippocampus Controls Spatial Memory Deficits in a Mouse Model of Autism and Intellectual Disability. Biol. Psychiatry 2019, 85, 760–768. [Google Scholar] [CrossRef]
- Shao, L.; Lu, B.; Wen, Z.; Teng, S.; Wang, L.; Zhao, Y.; Wang, L.; Ishizuka, K.; Xu, X.; Sawa, A.; et al. Disrupted-in-Schizophrenia-1 (DISC1) protein disturbs neural function in multiple disease-risk pathways. Hum. Mol. Genet. 2017, 26, 2634–2648. [Google Scholar] [CrossRef]
- Thomson, P.A.; Malavasi, E.L.; Grünewald, E.; Soares, D.C.; Borkowska, M.; Millar, J.K. DISC1 genetics, biology and psychiatric illness. Front. Biol. 2013, 8, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Kang, E.; Yu, C.; Qian, X.; Jacob, F.; Yu, C.; Mao, M.; Poon, R.Y.C.; Kim, J.; Song, H.; et al. DISC1 Regulates Neurogenesis via Modulating Kinetochore Attachment of Ndel1/Nde1 during Mitosis. Neuron 2017, 96, 1041–1054. [Google Scholar] [CrossRef]
- Kvajo, M.; McKellar, H.; Arguello, P.A.; Drew, L.J.; Moore, H.; MacDermott, A.B.; Karayiorgou, M.; Gogos, J.A. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. USA 2008, 105, 7076–7081. [Google Scholar] [CrossRef]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Frank, C.; Ishizuka, K.; Baccarella, A.; Park, Y.U.; Flood, Z.; Park, S.K.; Sawa, A.; Tsai, L.H. DISC1-ATF4 transcriptional repression complex: Dual regulation of the cAMP-PDE4 cascade by DISC1. Mol. Psychiatry 2013, 18, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, N.; Ando, T.; Maruyama, Y.; Fujimuro, M.; Mochizuki, H.; Honjo, K.; Shimoda, M.; Toda, H.; Sawamura-Yamamoto, T.; Makuch, L.A.; et al. Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Mol. Psychiatry 2008, 13, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Ukkola-Vuoti, L.; Ortega-Alonso, A.; Torniainen-Holm, M.; Therman, S.; Tuulio-Henriksson, A.; Jylhä, P.; Kaprio, J.; Hovatta, I.; Isometsä, E.; et al. Variants in regulatory elements of PDE4D associate with major mental illness in the Finnish population. Mol. Psychiatry 2021, 26, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Green, T.A.; Alibhai, I.N.; Unterberg, S.; Neve, R.L.; Ghose, S.; Tamminga, C.A.; Nestler, E.J. Induction of activating transcription factors (ATFs) ATF2, ATF3, and ATF4 in the nucleus accumbens and their regulation of emotional behavior. J. Neurosci. 2008, 28, 2025–2032. [Google Scholar] [CrossRef]
- Kuroiwa, M.; Snyder, G.L.; Shuto, T.; Fukuda, A.; Yanagawa, Y.; Benavides, D.R.; Nairn, A.C.; Bibb, J.A.; Greengard, P.; Nishi, A. Phosphodiesterase 4 inhibition enhances the dopamine D1 receptor/PKA/DARPP-32 signaling cascade in frontal cortex. Psychopharmacology 2012, 219, 1065–1079. [Google Scholar] [CrossRef]
- Su, P.; Zhang, H.; Wong, A.H.C.; Liu, F. The DISC1 R264Q variant increases affinity for the dopamine D2 receptor and increases GSK3 activity. Mol. Brain 2020, 13, 87. [Google Scholar] [CrossRef]
- Kim, N.S.; Wen, Z.; Liu, J.; Zhou, Y.; Guo, Z.; Xu, C.; Lin, Y.T.; Yoon, K.J.; Park, J.; Cho, M.; et al. Pharmacological rescue in patient iPSC and mouse models with a rare DISC1 mutation. Nat. Commun. 2021, 12, 1398. [Google Scholar] [CrossRef]
- Millar, J.K.; Pickard, B.S.; Mackie, S.; James, R.; Christie, S.; Buchanan, S.R.; Malloy, M.P.; Chubb, J.E.; Huston, E.; Baillie, G.S.; et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 2005, 310, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.S.; Yoon, K.J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Pearse, D.D.; Pereira, F.C.; Marcillo, A.E.; Bates, M.L.; Berrocal, Y.A.; Filbin, M.T.; Bunge, M.B. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 2004, 10, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Su, S.C.; Gao, J.; Joseph, N.; Xie, Z.; Zhou, Y.; Durak, O.; Zhang, L.; Zhu, J.J.; Clauser, K.R.; et al. Cdk5 is required for memory function and hippocampal plasticity via the cAMP signaling pathway. PLoS ONE 2011, 6, e25735. [Google Scholar] [CrossRef]
- Bradshaw, N.J.; Porteous, D.J. DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology 2012, 62, 1230–1241. [Google Scholar] [CrossRef]
- Song, W.; Li, W.; Feng, J.; Heston, L.L.; Scaringe, W.A.; Sommer, S.S. Identification of high risk DISC1 structural variants with a 2% attributable risk for schizophrenia. Biochem. Biophys. Res. Commun. 2008, 367, 700–706. [Google Scholar] [CrossRef]
- Song, W.; Li, W.; Noltner, K.; Yan, J.; Green, E.; Grozeva, D.; Jones, I.R.; Craddock, N.; Longmate, J.; Feng, J.; et al. Identification of high risk DISC1 protein structural variants in patients with bipolar spectrum disorder. Neurosci. Lett. 2010, 486, 136–140. [Google Scholar] [CrossRef]
- Green, E.K.; Grozeva, D.; Sims, R.; Raybould, R.; Forty, L.; Gordon-Smith, K.; Russell, E.; St Clair, D.; Young, A.H.; Ferrier, I.N.; et al. DISC1 exon 11 rare variants found more commonly in schizoaffective spectrum cases than controls. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156b, 490–492. [Google Scholar] [CrossRef]
- Brauns, S.; Gollub, R.L.; Roffman, J.L.; Yendiki, A.; Ho, B.C.; Wassink, T.H.; Heinz, A.; Ehrlich, S. DISC1 is associated with cortical thickness and neural efficiency. NeuroImage 2011, 57, 1591–1600. [Google Scholar] [CrossRef]
- Szeszko, P.R.; Hodgkinson, C.A.; Robinson, D.G.; Derosse, P.; Bilder, R.M.; Lencz, T.; Burdick, K.E.; Napolitano, B.; Betensky, J.D.; Kane, J.M.; et al. DISC1 is associated with prefrontal cortical gray matter and positive symptoms in schizophrenia. Biol. Psychol. 2008, 79, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Iasevoli, F.; D’Ambrosio, L.; Ciccarelli, M.; Barone, A.; Gaudieri, V.; Cocozza, S.; Pontillo, G.; Brunetti, A.; Cuocolo, A.; de Bartolomeis, A.; et al. Altered Patterns of Brain Glucose Metabolism Involve More Extensive and Discrete Cortical Areas in Treatment-resistant Schizophrenia Patients Compared to Responder Patients and Controls: Results From a Head-to-Head 2-[18F]-FDG-PET Study. Schizophr. Bull. 2022, 147. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, S.L.; Hodgkinson, C.A.; Harrison, P.J. DISC-1 Leu607Phe alleles differentially affect centrosomal PCM1 localization and neurotransmitter release. Mol. Psychiatry 2009, 14, 556–557. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Lipska, B.K.; Hyde, T.M.; Ye, T.; Newburn, E.N.; Morita, Y.; Vakkalanka, R.; Barenboim, M.; Sei, Y.; Weinberger, D.R.; et al. DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proc. Natl. Acad. Sci. USA 2009, 106, 15873–15878. [Google Scholar] [CrossRef]
- Hashimoto, R.; Numakawa, T.; Ohnishi, T.; Kumamaru, E.; Yagasaki, Y.; Ishimoto, T.; Mori, T.; Nemoto, K.; Adachi, N.; Izumi, A.; et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum. Mol. Genet. 2006, 15, 3024–3033. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Suzuki, M.; Tsunoda, M.; Maeno, N.; Kawasaki, Y.; Zhou, S.Y.; Hagino, H.; Niu, L.; Tsuneki, H.; Kobayashi, S.; et al. The Disrupted-in-Schizophrenia-1 Ser704Cys polymorphism and brain morphology in schizophrenia. Psychiatry Res. 2009, 172, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, A.; Blasi, G.; Sambataro, F.; Rampino, A.; Papazacharias, A.; Gambi, F.; Romano, R.; Caforio, G.; Rizzo, M.; Latorre, V.; et al. Association of the SerCys DISC1 polymorphism with human hippocampal formation gray matter and function during memory encoding. Eur. J. Neurosci. 2008, 28, 2129–2136. [Google Scholar] [CrossRef]
- DeRosse, P.; Hodgkinson, C.A.; Lencz, T.; Burdick, K.E.; Kane, J.M.; Goldman, D.; Malhotra, A.K. Disrupted in schizophrenia 1 genotype and positive symptoms in schizophrenia. Biol. Psychiatry 2007, 61, 1208–1210. [Google Scholar] [CrossRef]
- Prata, D.P.; Mechelli, A.; Fu, C.H.; Picchioni, M.; Kane, F.; Kalidindi, S.; McDonald, C.; Kravariti, E.; Toulopoulou, T.; Miorelli, A.; et al. Effect of disrupted-in-schizophrenia-1 on pre-frontal cortical function. Mol. Psychiatry 2008, 13, 915–917. [Google Scholar] [CrossRef]
- Eastwood, S.L.; Walker, M.; Hyde, T.M.; Kleinman, J.E.; Harrison, P.J. The DISC1 Ser704Cys substitution affects centrosomal localization of its binding partner PCM1 in glia in human brain. Hum. Mol. Genet. 2010, 19, 2487–2496. [Google Scholar] [CrossRef]
- Leliveld, S.R.; Hendriks, P.; Michel, M.; Sajnani, G.; Bader, V.; Trossbach, S.; Prikulis, I.; Hartmann, R.; Jonas, E.; Willbold, D.; et al. Oligomer assembly of the C-terminal DISC1 domain (640-854) is controlled by self-association motifs and disease-associated polymorphism S704C. Biochemistry 2009, 48, 7746–7755. [Google Scholar] [CrossRef]
- Burdick, K.E.; Kamiya, A.; Hodgkinson, C.A.; Lencz, T.; DeRosse, P.; Ishizuka, K.; Elashvili, S.; Arai, H.; Goldman, D.; Sawa, A.; et al. Elucidating the relationship between DISC1, NDEL1 and NDE1 and the risk for schizophrenia: Evidence of epistasis and competitive binding. Hum. Mol. Genet. 2008, 17, 2462–2473. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, M.; Thomson, P.A.; Hall, J.; McIntosh, A.M.; Lawrie, S.M.; Porteous, D.J. DISC1 in schizophrenia: Genetic mouse models and human genomic imaging. Schizophr. Bull. 2011, 37, 14–20. [Google Scholar] [CrossRef]
- Wang, Q.; Jaaro-Peled, H.; Sawa, A.; Brandon, N.J. How has DISC1 enabled drug discovery? Mol. Cell. Neurosci. 2008, 37, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Hashimoto, R.; Hattori, S.; Yohda, M.; Lipska, B.; Weinberger, D.R.; Kunugi, H. Effect of antipsychotic drugs on DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J. Neural Transm. 2006, 113, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Mouaffak, F.; Kebir, O.; Chayet, M.; Tordjman, S.; Vacheron, M.N.; Millet, B.; Jaafari, N.; Bellon, A.; Olié, J.P.; Krebs, M.O. Association of Disrupted in Schizophrenia 1 (DISC1) missense variants with ultra-resistant schizophrenia. Pharm. J. 2011, 11, 267–273. [Google Scholar] [CrossRef]
- Nagai, T.; Kitahara, Y.; Ibi, D.; Nabeshima, T.; Sawa, A.; Yamada, K. Effects of antipsychotics on the behavioral deficits in human dominant-negative DISC1 transgenic mice with neonatal polyI:C treatment. Behav. Brain Res. 2011, 225, 305–310. [Google Scholar] [CrossRef]
- Brakeman, P.R.; Lanahan, A.A.; O’Brien, R.; Roche, K.; Barnes, C.A.; Huganir, R.L.; Worley, P.F. Homer: A protein that selectively binds metabotropic glutamate receptors. Nature 1997, 386, 284–288. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Iasevoli, F. The Homer family and the signal transduction system at glutamatergic postsynaptic density: Potential role in behavior and pharmacotherapy. Psychopharmacol. Bull. 2003, 37, 51–83. [Google Scholar]
- de Bartolomeis, A.; Barone, A.; Vellucci, L.; Mazza, B.; Austin, M.C.; Iasevoli, F.; Ciccarelli, M. Linking Inflammation, Aberrant Glutamate-Dopamine Interaction, and Post-synaptic Changes: Translational Relevance for Schizophrenia and Antipsychotic Treatment: A Systematic Review. Mol. Neurobiol. 2022, 59, 6460–6501. [Google Scholar] [CrossRef]
- Soloviev, M.M.; Ciruela, F.; Chan, W.Y.; McIlhinney, R.A. Mouse brain and muscle tissues constitutively express high levels of Homer proteins. Eur. J. Biochem. 2000, 267, 634–639. [Google Scholar] [CrossRef]
- Blottner, D.; Trautmann, G.; Furlan, S.; Gambara, G.; Block, K.; Gutsmann, M.; Sun, L.W.; Worley, P.F.; Gorza, L.; Scano, M.; et al. Reciprocal Homer1a and Homer2 Isoform Expression Is a Key Mechanism for Muscle Soleus Atrophy in Spaceflown Mice. Int. J. Mol. Sci. 2021, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, L.; Shi, C.; Wei, R.; Yin, C. Interaction of the Homer1 EVH1 domain and skeletal muscle ryanodine receptor. Biochem. Biophys. Res. Commun. 2019, 514, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Imamura, N.; Nonaka, A.; Yamamoto, H.; Matsuki, N.; Nomura, H. Experience-dependent Homer1a expression in excitatory and inhibitory neurons. Neuroreport 2011, 22, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.C.; Xiao, B.; Yuan, J.P.; Lanahan, A.A.; Leoffert, K.; Li, M.; Linden, D.J.; Worley, P.F. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 1998, 21, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Tappe, A.; Kuner, R. Regulation of motor performance and striatal function by synaptic scaffolding proteins of the Homer1 family. Proc. Natl. Acad. Sci. USA 2006, 103, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Gimse, K.; Gorzek, R.C.; Olin, A.; Osting, S.; Burger, C. Hippocampal Homer1b/c is necessary for contextual fear conditioning and group I metabotropic glutamate receptor mediated long-term depression. Neurobiol. Learn. Mem. 2018, 156, 17–23. [Google Scholar] [CrossRef]
- Celikel, T.; Marx, V.; Freudenberg, F.; Zivkovic, A.; Resnik, E.; Hasan, M.T.; Licznerski, P.; Osten, P.; Rozov, A.; Seeburg, P.H.; et al. Select overexpression of homer1a in dorsal hippocampus impairs spatial working memory. Front. Neurosci. 2007, 1, 97–110. [Google Scholar] [CrossRef]
- Lominac, K.D.; Oleson, E.B.; Pava, M.; Klugmann, M.; Schwarz, M.K.; Seeburg, P.H.; During, M.J.; Worley, P.F.; Kalivas, P.W.; Szumlinski, K.K. Distinct roles for different Homer1 isoforms in behaviors and associated prefrontal cortex function. J. Neurosci. 2005, 25, 11586–11594. [Google Scholar] [CrossRef]
- Rietschel, M.; Mattheisen, M.; Frank, J.; Treutlein, J.; Degenhardt, F.; Breuer, R.; Steffens, M.; Mier, D.; Esslinger, C.; Walter, H.; et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol. Psychiatry 2010, 68, 578–585. [Google Scholar] [CrossRef]
- Robbins, T.W.; Arnsten, A.F. The neuropsychopharmacology of fronto-executive function: Monoaminergic modulation. Annu. Rev. Neurosci. 2009, 32, 267–287. [Google Scholar] [CrossRef]
- Simpson, E.H.; Kellendonk, C.; Kandel, E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 2010, 65, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Tamminga, C.A. The neurobiology of cognition in schizophrenia. J. Clin. Psychiatry 2006, 67 (Suppl. 9), 9–13. [Google Scholar] [CrossRef] [PubMed]
- Stillman, M.; Lautz, J.D.; Johnson, R.S.; MacCoss, M.J.; Smith, S.E.P. Activity dependent dissociation of the Homer1 interactome. Sci. Rep. 2022, 12, 3207. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Bridi, M.; Schoch, H.; Florian, C.; Poplawski, S.G.; Banerjee, A.; Hawk, J.D.; Porcari, G.S.; Lejards, C.; Hahn, C.G.; Giese, K.P.; et al. Transcriptional corepressor SIN3A regulates hippocampal synaptic plasticity via Homer1/mGluR5 signaling. JCI Insight 2020, 5, e92385. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, V.; Gao, M.; Grier, B.D.; Owens, A.; Wang, H.; Worley, P.F.; Lee, H.K. Input-Specific Metaplasticity in the Visual Cortex Requires Homer1a-Mediated mGluR5 Signaling. Neuron 2019, 104, 736–748.e6. [Google Scholar] [CrossRef]
- Yoon, S.; Piguel, N.H.; Khalatyan, N.; Dionisio, L.E.; Savas, J.N.; Penzes, P. Homer1 promotes dendritic spine growth through ankyrin-G and its loss reshapes the synaptic proteome. Mol. Psychiatry 2021, 26, 1775–1789. [Google Scholar] [CrossRef]
- Kang, J.; Park, H.; Kim, E. IRSp53/BAIAP2 in dendritic spine development, NMDA receptor regulation, and psychiatric disorders. Neuropharmacology 2016, 100, 27–39. [Google Scholar] [CrossRef]
- Worley, P.F.; Zeng, W.; Huang, G.; Kim, J.Y.; Shin, D.M.; Kim, M.S.; Yuan, J.P.; Kiselyov, K.; Muallem, S. Homer proteins in Ca2+ signaling by excitable and non-excitable cells. Cell Calcium 2007, 42, 363–371. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Mizutani, A.; Mikoshiba, K.; Furuichi, T. Coincidence in dendritic clustering and synaptic targeting of homer proteins and NMDA receptor complex proteins NR2B and PSD95 during development of cultured hippocampal neurons. Mol. Cell. Neurosci. 2003, 22, 188–201. [Google Scholar] [CrossRef]
- Norton, N.; Williams, H.J.; Williams, N.M.; Spurlock, G.; Zammit, S.; Jones, G.; Jones, S.; Owen, R.; O’Donovan, M.C.; Owen, M.J. Mutation screening of the Homer gene family and association analysis in schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2003, 120, 18–21. [Google Scholar] [CrossRef]
- Shelley, C.; Farrant, M.; Cull-Candy, S.G. TARP-associated AMPA receptors display an increased maximum channel conductance and multiple kinetically distinct open states. J. Physiol. 2012, 590, 5723–5738. [Google Scholar] [CrossRef] [PubMed]
- Louros, S.R.; Caldeira, G.L.; Carvalho, A.L. Stargazin Dephosphorylation Mediates Homeostatic Synaptic Downscaling of Excitatory Synapses. Front. Mol. Neurosci. 2018, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yaacov, A.; Gillor, M.; Haham, T.; Parsai, A.; Qneibi, M.; Stern-Bach, Y. Molecular Mechanism of AMPA Receptor Modulation by TARP/Stargazin. Neuron 2017, 93, 1126–1137.e1. [Google Scholar] [CrossRef]
- Tomita, S.; Adesnik, H.; Sekiguchi, M.; Zhang, W.; Wada, K.; Howe, J.R.; Nicoll, R.A.; Bredt, D.S. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 2005, 435, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Schnell, E.; Sizemore, M.; Karimzadegan, S.; Chen, L.; Bredt, D.S.; Nicoll, R.A. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc. Natl. Acad. Sci. USA 2002, 99, 13902–13907. [Google Scholar] [CrossRef]
- Sturgill, J.F.; Steiner, P.; Czervionke, B.L.; Sabatini, B.L. Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J. Neurosci. 2009, 29, 12845–12854. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Payne, H.L. The role of transmembrane AMPA receptor regulatory proteins (TARPs) in neurotransmission and receptor trafficking (Review). Mol. Membr. Biol. 2008, 25, 353–362. [Google Scholar] [CrossRef]
- Migaud, M.; Charlesworth, P.; Dempster, M.; Webster, L.C.; Watabe, A.M.; Makhinson, M.; He, Y.; Ramsay, M.F.; Morris, R.G.; Morrison, J.H.; et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 1998, 396, 433–439. [Google Scholar] [CrossRef]
- Benesh, J.L.; Mueller, T.M.; Meador-Woodruff, J.H. AMPA receptor subunit localization in schizophrenia anterior cingulate cortex. Schizophr. Res. 2022, 249, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Maher, M.P.; Wu, N.; Ravula, S.; Ameriks, M.K.; Savall, B.M.; Liu, C.; Lord, B.; Wyatt, R.M.; Matta, J.A.; Dugovic, C.; et al. Discovery and Characterization of AMPA Receptor Modulators Selective for TARP-γ8. J. Pharmacol. Exp. Ther. 2016, 357, 394–414. [Google Scholar] [CrossRef] [PubMed]
- Ravula, S.; Savall, B.M.; Wu, N.; Lord, B.; Coe, K.; Wang, K.; Seierstad, M.; Swanson, D.M.; Ziff, J.; Nguyen, M.; et al. Lead Optimization of 5-Aryl Benzimidazolone- and Oxindole-Based AMPA Receptor Modulators Selective for TARP γ-8. ACS Med. Chem. Lett. 2018, 9, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Shekhtman, T.; McCarthy, M.; DeModena, A.; Leckband, S.G.; Kelsoe, J.R. Study of 45 candidate genes suggests CACNG2 may be associated with lithium response in bipolar disorder. J. Affect. Disord. 2019, 248, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.S.; Gill, M.B.; Yu, H.; Nisenbaum, E.S.; Bredt, D.S. TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010, 33, 241–248. [Google Scholar] [CrossRef]
- Soler, J.; Fañanás, L.; Parellada, M.; Krebs, M.O.; Rouleau, G.A.; Fatjó-Vilas, M. Genetic variability in scaffolding proteins and risk for schizophrenia and autism-spectrum disorders: A systematic review. J. Psychiatry Neurosci. 2018, 43, 223–244. [Google Scholar] [CrossRef]
- Leber, S.L.; Llenos, I.C.; Miller, C.L.; Dulay, J.R.; Haybaeck, J.; Weis, S. Homer1a protein expression in schizophrenia, bipolar disorder, and major depression. J. Neural Transm. 2017, 124, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R.; Alme, M.N.; Bittins, M.; Kuipers, S.D.; Nair, R.R.; Pai, B.; Panja, D.; Schubert, M.; Soule, J.; Tiron, A.; et al. The Arc of synaptic memory. Exp. Brain Res. 2010, 200, 125–140. [Google Scholar] [CrossRef]
- Campillos, M.; Doerks, T.; Shah, P.K.; Bork, P. Computational characterization of multiple Gag-like human proteins. Trends Genet. 2006, 22, 585–589. [Google Scholar] [CrossRef]
- Ashley, J.; Cordy, B.; Lucia, D.; Fradkin, L.G.; Budnik, V.; Thomson, T. Retrovirus-like Gag Protein Arc1 Binds RNA and Traffics across Synaptic Boutons. Cell 2018, 172, 262–274.e2. [Google Scholar] [CrossRef]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The Neuronal Gene Arc Encodes a Repurposed Retrotransposon Gag Protein that Mediates Intercellular RNA Transfer. Cell 2018, 172, 275–288.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fila, M.; Diaz, L.; Szczepanska, J.; Pawlowska, E.; Blasiak, J. mRNA Trafficking in the Nervous System: A Key Mechanism of the Involvement of Activity-Regulated Cytoskeleton-Associated Protein (Arc) in Synaptic Plasticity. Neural Plast. 2021, 2021, 3468795. [Google Scholar] [CrossRef] [PubMed]
- Raju, C.S.; Fukuda, N.; López-Iglesias, C.; Göritz, C.; Visa, N.; Percipalle, P. In neurons, activity-dependent association of dendritically transported mRNA transcripts with the transacting factor CBF-A is mediated by A2RE/RTS elements. Mol. Biol. Cell 2011, 22, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Okuno, H.; Akashi, K.; Ishii, Y.; Yagishita-Kyo, N.; Suzuki, K.; Nonaka, M.; Kawashima, T.; Fujii, H.; Takemoto-Kimura, S.; Abe, M.; et al. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIβ. Cell 2012, 149, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Lyford, G.L.; Yamagata, K.; Kaufmann, W.E.; Barnes, C.A.; Sanders, L.K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Lanahan, A.A.; Worley, P.F. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 1995, 14, 433–445. [Google Scholar] [CrossRef]
- Steward, O.; Worley, P.F. A cellular mechanism for targeting newly synthesized mRNAs to synaptic sites on dendrites. Proc. Natl. Acad. Sci. USA 2001, 98, 7062–7068. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Amaya, V.; Vazdarjanova, A.; Mikhael, D.; Rosi, S.; Worley, P.F.; Barnes, C.A. Spatial exploration-induced Arc mRNA and protein expression: Evidence for selective, network-specific reactivation. J. Neurosci. 2005, 25, 1761–1768. [Google Scholar] [CrossRef]
- Korb, E.; Finkbeiner, S. Arc in synaptic plasticity: From gene to behavior. Trends Neurosci. 2011, 34, 591–598. [Google Scholar] [CrossRef]
- Rao, V.R.; Pintchovski, S.A.; Chin, J.; Peebles, C.L.; Mitra, S.; Finkbeiner, S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat. Neurosci. 2006, 9, 887–895. [Google Scholar] [CrossRef]
- Fujimoto, T.; Tanaka, H.; Kumamaru, E.; Okamura, K.; Miki, N. Arc interacts with microtubules/microtubule-associated protein 2 and attenuates microtubule-associated protein 2 immunoreactivity in the dendrites. J. Neurosci. Res. 2004, 76, 51–63. [Google Scholar] [CrossRef]
- Messaoudi, E.; Kanhema, T.; Soulé, J.; Tiron, A.; Dagyte, G.; da Silva, B.; Bramham, C.R. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J. Neurosci. 2007, 27, 10445–10455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peebles, C.L.; Yoo, J.; Thwin, M.T.; Palop, J.J.; Noebels, J.L.; Finkbeiner, S. Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 18173–18178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pehrson, A.L.; Waller, J.A.; Dale, E.; Sanchez, C.; Gulinello, M. A critical evaluation of the activity-regulated cytoskeleton-associated protein (Arc/Arg3.1)’s putative role in regulating dendritic plasticity, cognitive processes, and mood in animal models of depression. Front. Neurosci. 2015, 9, 279. [Google Scholar] [CrossRef]
- Chowdhury, S.; Shepherd, J.D.; Okuno, H.; Lyford, G.; Petralia, R.S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 2006, 52, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Rial Verde, E.M.; Lee-Osbourne, J.; Worley, P.F.; Malinow, R.; Cline, H.T. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron 2006, 52, 461–474. [Google Scholar] [CrossRef]
- Waung, M.W.; Pfeiffer, B.E.; Nosyreva, E.D.; Ronesi, J.A.; Huber, K.M. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 2008, 59, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Korb, E.; Wilkinson, C.L.; Delgado, R.N.; Lovero, K.L.; Finkbeiner, S. Arc in the nucleus regulates PML-dependent GluA1 transcription and homeostatic plasticity. Nat. Neurosci. 2013, 16, 874–883. [Google Scholar] [CrossRef]
- Steward, O.; Worley, P.F. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 2001, 30, 227–240. [Google Scholar] [CrossRef]
- Panja, D.; Dagyte, G.; Bidinosti, M.; Wibrand, K.; Kristiansen, A.M.; Sonenberg, N.; Bramham, C.R. Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J. Biol. Chem. 2009, 284, 31498–31511. [Google Scholar] [CrossRef]
- Crisafulli, C.; Calabrò, M.; Mandelli, L.; Wang, S.M.; Lee, S.J.; Han, C.; Patkar, A.; Masand, P.; Pae, C.U.; Souery, D.; et al. Possible Modulatory Role of ARC Gene Variants in Mood Disorders. Clin. Psychopharmacol. Neurosci. 2021, 19, 46–52. [Google Scholar] [CrossRef]
- Hou, W.L.; Yin, X.L.; Yin, X.Y.; Guan, L.Y.; Cao, J.Q.; Tang, Z.; Jiang, C.X.; Xu, D.W.; Yu, X.; Wang, J.; et al. Association between stereopsis deficits and attention decline in patients with major depressive disorder. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2021, 110, 110267. [Google Scholar] [CrossRef] [PubMed]
- Pine, J.G.; Moe, A.M.; Maple, A.M.; Gallitano, A.L.; Breitborde, N.J.K. Activity-regulated cytoskeleton-associated protein predicts symptom response to cognitive behavioral therapy among individuals with first-episode psychosis. Asian J. Psychiatry 2020, 50, 101974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, J.; Ward, M.D.; Yang, S.; Chuang, Y.A.; Xiao, M.; Li, R.; Leahy, D.J.; Worley, P.F. Structural basis of arc binding to synaptic proteins: Implications for cognitive disease. Neuron 2015, 86, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef]
- Bi, R.; Kong, L.L.; Xu, M.; Li, G.D.; Zhang, D.F.; Li, T.; Fang, Y.; Zhang, C.; Zhang, B.; Yao, Y.G. The Arc Gene Confers Genetic Susceptibility to Alzheimer’s Disease in Han Chinese. Mol. Neurobiol. 2018, 55, 1217–1226. [Google Scholar] [CrossRef]
- Landgren, S.; von Otter, M.; Palmér, M.S.; Zetterström, C.; Nilsson, S.; Skoog, I.; Gustafson, D.R.; Minthon, L.; Wallin, A.; Andreasen, N.; et al. A novel ARC gene polymorphism is associated with reduced risk of Alzheimer’s disease. J. Neural Transm. 2012, 119, 833–842. [Google Scholar] [CrossRef]
- Postu, P.A.; Tiron, A.; Tiron, C.E.; Gorgan, D.L.; Mihasan, M.; Hritcu, L. Conifer Essential Oils Reversed Amyloid Beta1-42 Action by Modulating BDNF and ARC Expression in The Rat Hippocampus. CNS Neurol. Disord. Drug Targets 2022, 21, 85–94. [Google Scholar] [CrossRef]
- Wu, J.; Petralia, R.S.; Kurushima, H.; Patel, H.; Jung, M.Y.; Volk, L.; Chowdhury, S.; Shepherd, J.D.; Dehoff, M.; Li, Y.; et al. Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent β-amyloid generation. Cell 2011, 147, 615–628. [Google Scholar] [CrossRef]
- Shepherd, J.D.; Bear, M.F. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 2011, 14, 279–284. [Google Scholar] [CrossRef]
- Managò, F.; Mereu, M.; Mastwal, S.; Mastrogiacomo, R.; Scheggia, D.; Emanuele, M.; De Luca, M.A.; Weinberger, D.R.; Wang, K.H.; Papaleo, F. Genetic Disruption of Arc/Arg3.1 in Mice Causes Alterations in Dopamine and Neurobehavioral Phenotypes Related to Schizophrenia. Cell Rep. 2016, 16, 2116–2128. [Google Scholar] [CrossRef]
- Kitamura, S.; Kuratomi, Y.; Aoki, S.; Ohno, S. Change of blood levels of leukotrienes and thromboxane B2 induced by anaphylactic shock in anesthetized dogs. Adv. Prostaglandin Thromboxane Leukot. Res. 1987, 17b, 1038–1042. [Google Scholar] [PubMed]
- Bruins Slot, L.A.; Lestienne, F.; Grevoz-Barret, C.; Newman-Tancredi, A.; Cussac, D. F15063, a potential antipsychotic with dopamine D(2)/D(3) receptor antagonist and 5-HT(1A) receptor agonist properties: Influence on immediate-early gene expression in rat prefrontal cortex and striatum. Eur. J. Pharmacol. 2009, 620, 27–35. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Marmo, F.; Buonaguro, E.F.; Rossi, R.; Tomasetti, C.; Iasevoli, F. Imaging brain gene expression profiles by antipsychotics: Region-specific action of amisulpride on postsynaptic density transcripts compared to haloperidol. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2013, 23, 1516–1529. [Google Scholar] [CrossRef]
- Robbins, M.J.; Critchlow, H.M.; Lloyd, A.; Cilia, J.; Clarke, J.D.; Bond, B.; Jones, D.N.; Maycox, P.R. Differential expression of IEG mRNA in rat brain following acute treatment with clozapine or haloperidol: A semi-quantitative RT-PCR study. J. Psychopharmacol. 2008, 22, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.M.; Wood, M.D.; Elliott, J.M. Chronic administration of haloperidol and clozapine induces differential effects on the expression of Arc and c-Fos in rat brain. J. Psychopharmacol. 2014, 28, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Frasca, A.; Racagni, G.; Riva, M.A. Antipsychotic drugs modulate Arc expression in the rat brain. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2009, 19, 109–115. [Google Scholar] [CrossRef]
- Nakahara, T.; Kuroki, T.; Hashimoto, K.; Hondo, H.; Tsutsumi, T.; Motomura, K.; Ueki, H.; Hirano, M.; Uchimura, H. Effect of atypical antipsychotics on phencyclidine-induced expression of arc in rat brain. Neuroreport 2000, 11, 551–555. [Google Scholar] [CrossRef]
- Fisahn, A.; Neddens, J.; Yan, L.; Buonanno, A. Neuregulin-1 modulates hippocampal gamma oscillations: Implications for schizophrenia. Cereb. Cortex 2009, 19, 612–618. [Google Scholar] [CrossRef]
- Wen, L.; Lu, Y.S.; Zhu, X.H.; Li, X.M.; Woo, R.S.; Chen, Y.J.; Yin, D.M.; Lai, C.; Terry, A.V., Jr.; Vazdarjanova, A.; et al. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. USA 2010, 107, 1211–1216. [Google Scholar] [CrossRef]
- Shi, L.; Bergson, C.M. Neuregulin 1: An intriguing therapeutic target for neurodevelopmental disorders. Transl. Psychiatry 2020, 10, 190. [Google Scholar] [CrossRef]
- Gu, Y.; Tran, T.; Murase, S.; Borrell, A.; Kirkwood, A.; Quinlan, E.M. Neuregulin-Dependent Regulation of Fast-Spiking Interneuron Excitability Controls the Timing of the Critical Period. J. Neurosci. 2016, 36, 10285–10295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, R.S.; Li, X.M.; Tao, Y.; Carpenter-Hyland, E.; Huang, Y.Z.; Weber, J.; Neiswender, H.; Dong, X.P.; Wu, J.; Gassmann, M.; et al. Neuregulin-1 enhances depolarization-induced GABA release. Neuron 2007, 54, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Eilam, R.; Pinkas-Kramarski, R.; Ratzkin, B.J.; Segal, M.; Yarden, Y. Activity-dependent regulation of Neu differentiation factor/neuregulin expression in rat brain. Proc. Natl. Acad. Sci. USA 1998, 95, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.S.; Mott, D.D. Neuregulin blocks synaptic strengthening after epileptiform activity in the rat hippocampus. Brain Res. 2008, 1208, 67–73. [Google Scholar] [CrossRef]
- Tan, G.H.; Liu, Y.Y.; Hu, X.L.; Yin, D.M.; Mei, L.; Xiong, Z.Q. Neuregulin 1 represses limbic epileptogenesis through ErbB4 in parvalbumin-expressing interneurons. Nat. Neurosci. 2011, 15, 258–266. [Google Scholar] [CrossRef]
- Tino, J.A.; Clark, J.M.; Field, A.K.; Jacobs, G.A.; Lis, K.A.; Michalik, T.L.; McGeever-Rubin, B.; Slusarchyk, W.A.; Spergel, S.H.; Sundeen, J.E.; et al. Synthesis and antiviral activity of novel isonucleoside analogs. J. Med. Chem. 1993, 36, 1221–1229. [Google Scholar] [CrossRef]
- Kwon, O.B.; Longart, M.; Vullhorst, D.; Hoffman, D.A.; Buonanno, A. Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. J. Neurosci. 2005, 25, 9378–9383. [Google Scholar] [CrossRef]
- Shamir, A.; Kwon, O.B.; Karavanova, I.; Vullhorst, D.; Leiva-Salcedo, E.; Janssen, M.J.; Buonanno, A. The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J. Neurosci. 2012, 32, 2988–2997. [Google Scholar] [CrossRef]
- Chen, Y.J.; Zhang, M.; Yin, D.M.; Wen, L.; Ting, A.; Wang, P.; Lu, Y.S.; Zhu, X.H.; Li, S.J.; Wu, C.Y.; et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 21818–21823. [Google Scholar] [CrossRef]
- Tan, Z.; Robinson, H.L.; Yin, D.M.; Liu, Y.; Liu, F.; Wang, H.; Lin, T.W.; Xing, G.; Gan, L.; Xiong, W.C.; et al. Dynamic ErbB4 Activity in Hippocampal-Prefrontal Synchrony and Top-Down Attention in Rodents. Neuron 2018, 98, 380–393.e3. [Google Scholar] [CrossRef]
- Del Pino, I.; García-Frigola, C.; Dehorter, N.; Brotons-Mas, J.R.; Alvarez-Salvado, E.; Martínez de Lagrán, M.; Ciceri, G.; Gabaldón, M.V.; Moratal, D.; Dierssen, M.; et al. Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 2013, 79, 1152–1168. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Woo, R.S.; Mei, L.; Malinow, R. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 2007, 54, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ikrar, T.; Davis, M.F.; Gong, N.; Zheng, X.; Luo, Z.D.; Lai, C.; Mei, L.; Holmes, T.C.; Gandhi, S.P.; et al. Neuregulin-1/ErbB4 Signaling Regulates Visual Cortical Plasticity. Neuron 2016, 92, 160–173. [Google Scholar] [CrossRef]
- Huang, Y.Z.; Won, S.; Ali, D.W.; Wang, Q.; Tanowitz, M.; Du, Q.S.; Pelkey, K.A.; Yang, D.J.; Xiong, W.C.; Salter, M.W.; et al. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron 2000, 26, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Vasudevan, K.; Buonanno, A. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 3596–3601. [Google Scholar] [CrossRef] [PubMed]
- Corfas, G.; Roy, K.; Buxbaum, J.D. Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat. Neurosci. 2004, 7, 575–580. [Google Scholar] [CrossRef]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2005, 10, 40–68. [Google Scholar] [CrossRef]
- Cao, S.X.; Wen, C.X.; Sun, R.; Han, J.X.; Sun, Y.H.; Xu, X.X.; Li, X.M.; Lian, H. ErbB4 regulate extracellular dopamine through the p38 MAPK signaling pathway. Neurosci. Lett. 2021, 751, 135830. [Google Scholar] [CrossRef] [PubMed]
- Kushima, I.; Nakamura, Y.; Aleksic, B.; Ikeda, M.; Ito, Y.; Shiino, T.; Okochi, T.; Fukuo, Y.; Ujike, H.; Suzuki, M.; et al. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr. Bull. 2012, 38, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Yokosuka, T.; Hirakawa, H.; Ishihara, C.; Yasukawa, S.; Yamazaki, M.; Koseki, H.; Yoshida, H.; Saito, T. Clustering of CARMA1 through SH3-GUK domain interactions is required for its activation of NF-κB signalling. Nat. Commun. 2015, 6, 5555. [Google Scholar] [CrossRef]
- Zheng, C.Y.; Petralia, R.S.; Wang, Y.X.; Kachar, B.; Wenthold, R.J. SAP102 is a highly mobile MAGUK in spines. J. Neurosci. 2010, 30, 4757–4766. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.M.; Kistner, U.; Kindler, S.; Chung, W.J.; Kuhlendahl, S.; Fenster, S.D.; Lau, L.F.; Veh, R.W.; Huganir, R.L.; Gundelfinger, E.D.; et al. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron 1996, 17, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.A.; Elias, G.M.; Elias, L.A.; Swat, W.; Nicoll, R.A. The role of SAP97 in synaptic glutamate receptor dynamics. Proc. Natl. Acad. Sci. USA 2010, 107, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.Y.; Seabold, G.K.; Horak, M.; Petralia, R.S. MAGUKs, synaptic development, and synaptic plasticity. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2011, 17, 493–512. [Google Scholar] [CrossRef]
- Kim, J.H.; Liao, D.; Lau, L.F.; Huganir, R.L. SynGAP: A synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 1998, 20, 683–691. [Google Scholar] [CrossRef]
- Ting, Y.K.; Morikawa, K.; Kurata, Y.; Li, P.; Bahrudin, U.; Mizuta, E.; Kato, M.; Miake, J.; Yamamoto, Y.; Yoshida, A.; et al. Transcriptional activation of the anchoring protein SAP97 by heat shock factor (HSF)-1 stabilizes K(v) 1.5 channels in HL-1 cells. Br. J. Pharmacol. 2011, 162, 1832–1842. [Google Scholar] [CrossRef]
- Wu, H.; Nash, J.E.; Zamorano, P.; Garner, C.C. Interaction of SAP97 with minus-end-directed actin motor myosin VI. Implications for AMPA receptor trafficking. J. Biol. Chem. 2002, 277, 30928–30934. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Mauceri, D.; Fiorentini, C.; Bellone, C.; Missale, C.; Cattabeni, F.; Di Luca, M. CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J. Biol. Chem. 2003, 278, 44745–44752. [Google Scholar] [CrossRef]
- Sans, N.; Petralia, R.S.; Wang, Y.X.; Blahos, J., 2nd; Hell, J.W.; Wenthold, R.J. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J. Neurosci. 2000, 20, 1260–1271. [Google Scholar] [CrossRef]
- Gambrill, A.C.; Barria, A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. USA 2011, 108, 5855–5860. [Google Scholar] [CrossRef]
- Nikandrova, Y.A.; Jiao, Y.; Baucum, A.J.; Tavalin, S.J.; Colbran, R.J. Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J. Biol. Chem. 2010, 285, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbert, P.C.; Stanford, L.E.; Coba, M.P.; Ainge, J.A.; Fink, A.E.; Opazo, P.; Delgado, J.Y.; Komiyama, N.H.; O’Dell, T.J.; Grant, S.G. Synapse-associated protein 102/dlgh3 couples the NMDA receptor to specific plasticity pathways and learning strategies. J. Neurosci. 2007, 27, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Matus, A. Postsynaptic actin and neuronal plasticity. Curr. Opin. Neurobiol. 1999, 9, 561–565. [Google Scholar] [CrossRef]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef]
- Okabe, S.; Miwa, A.; Okado, H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J. Neurosci. 2001, 21, 6105–6114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Benson, D.L. Stages of synapse development defined by dependence on F-actin. J. Neurosci. 2001, 21, 5169–5181. [Google Scholar] [CrossRef]
- Furuyashiki, T.; Arakawa, Y.; Takemoto-Kimura, S.; Bito, H.; Narumiya, S. Multiple spatiotemporal modes of actin reorganization by NMDA receptors and voltage-gated Ca2+ channels. Proc. Natl. Acad. Sci. USA 2002, 99, 14458–14463. [Google Scholar] [CrossRef]
- Halpain, S.; Hipolito, A.; Saffer, L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. 1998, 18, 9835–9844. [Google Scholar] [CrossRef]
- Chen, C.C.; Lu, J.; Zuo, Y. Spatiotemporal dynamics of dendritic spines in the living brain. Front. Neuroanat. 2014, 8, 28. [Google Scholar] [CrossRef]
- Muller, D. Ultrastructural plasticity of excitatory synapses. Rev. Neurosci. 1997, 8, 77–93. [Google Scholar] [CrossRef]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, R.; Repetto, D.; Morini, R.; Elia, C.; Gardoni, F.; Di Luca, M.; Turco, E.; Defilippi, P.; Matteoli, M. SNAP-25 regulates spine formation through postsynaptic binding to p140Cap. Nat. Commun. 2013, 4, 2136. [Google Scholar] [CrossRef]
- Repetto, D.; Camera, P.; Melani, R.; Morello, N.; Russo, I.; Calcagno, E.; Tomasoni, R.; Bianchi, F.; Berto, G.; Giustetto, M.; et al. p140Cap regulates memory and synaptic plasticity through Src-mediated and citron-N-mediated actin reorganization. J. Neurosci. 2014, 34, 1542–1553. [Google Scholar] [CrossRef]
- Chin, L.S.; Nugent, R.D.; Raynor, M.C.; Vavalle, J.P.; Li, L. SNIP, a novel SNAP-25-interacting protein implicated in regulated exocytosis. J. Biol. Chem. 2000, 275, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Uruno, T.; Liu, J.; Zhang, P.; Fan, Y.; Egile, C.; Li, R.; Mueller, S.C.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef]
- Koch, E.; Rosenthal, B.; Lundquist, A.; Chen, C.H.; Kauppi, K. Interactome overlap between schizophrenia and cognition. Schizophr. Res. 2020, 222, 167–174. [Google Scholar] [CrossRef]
- Grintsevich, E.E. Effects of neuronal drebrin on actin dynamics. Biochem. Soc. Trans. 2021, 49, 685–692. [Google Scholar] [CrossRef]
- Aoki, C.; Sekino, Y.; Hanamura, K.; Fujisawa, S.; Mahadomrongkul, V.; Ren, Y.; Shirao, T. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J. Comp. Neurol. 2005, 483, 383–402. [Google Scholar] [CrossRef]
- Fujisawa, S.; Shirao, T.; Aoki, C. In vivo, competitive blockade of N-methyl-D-aspartate receptors induces rapid changes in filamentous actin and drebrin A distributions within dendritic spines of adult rat cortex. Neuroscience 2006, 140, 1177–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counts, S.E.; Nadeem, M.; Lad, S.P.; Wuu, J.; Mufson, E.J. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2006, 65, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Nakanishi, H.; Obaishi, H.; Wada, M.; Takahashi, K.; Satoh, K.; Hirao, K.; Nishioka, H.; Hata, Y.; Mizoguchi, A.; et al. Neurabin-II/spinophilin. An actin filament-binding protein with one pdz domain localized at cadherin-based cell-cell adhesion sites. J. Biol. Chem. 1998, 273, 3470–3475. [Google Scholar] [CrossRef]
- Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. USA 1997, 94, 9956–9961. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, C.C.; da Cruz e Silva, E.F.; Greengard, P. The alpha and gamma 1 isoforms of protein phosphatase 1 are highly and specifically concentrated in dendritic spines. Proc. Natl. Acad. Sci. USA 1995, 92, 3396–3400. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Orser, B.A.; Brautigan, D.L.; MacDonald, J.F. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature 1994, 369, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yan, Z.; Ferreira, A.; Tomizawa, K.; Liauw, J.A.; Zhuo, M.; Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. USA 2000, 97, 9287–9292. [Google Scholar] [CrossRef]
- Fischer, M.; Kaech, S.; Knutti, D.; Matus, A. Rapid actin-based plasticity in dendritic spines. Neuron 1998, 20, 847–854. [Google Scholar] [CrossRef]
- Fernandez, A.; Brautigan, D.L.; Mumby, M.; Lamb, N.J. Protein phosphatase type-1, not type-2A, modulates actin microfilament integrity and myosin light chain phosphorylation in living nonmuscle cells. J. Cell Biol. 1990, 111, 103–112. [Google Scholar] [CrossRef]
- Smith, F.D.; Oxford, G.S.; Milgram, S.L. Association of the D2 dopamine receptor third cytoplasmic loop with spinophilin, a protein phosphatase-1-interacting protein. J. Biol. Chem. 1999, 274, 19894–19900. [Google Scholar] [CrossRef]
- Obi-Nagata, K.; Temma, Y.; Hayashi-Takagi, A. Synaptic functions and their disruption in schizophrenia: From clinical evidence to synaptic optogenetics in an animal model. Proc. Jpn. Academy. Ser. B Phys. Biol. Sci. 2019, 95, 179–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennison, C.A.; Legge, S.E.; Pardiñas, A.F.; Walters, J.T.R. Genome-wide association studies in schizophrenia: Recent advances, challenges and future perspective. Schizophr. Res. 2020, 217, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Afia, A.B.; Vila, È.; MacDowell, K.S.; Ormazabal, A.; Leza, J.C.; Haro, J.M.; Artuch, R.; Ramos, B.; Garcia-Bueno, B. Kynurenine pathway in post-mortem prefrontal cortex and cerebellum in schizophrenia: Relationship with monoamines and symptomatology. J. Neuroinflammation 2021, 18, 198. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, H.; Kim, H.J.; Bang, E.; Lee, S.H.; Lee, D.W.; Woo, D.C.; Choi, C.B.; Hong, K.S.; Lee, C.; et al. In vivo and ex vivo evidence for ketamine-induced hyperglutamatergic activity in the cerebral cortex of the rat: Potential relevance to schizophrenia. NMR Biomed. 2011, 24, 1235–1242. [Google Scholar] [CrossRef]
- Koszła, O.; Targowska-Duda, K.M.; Kędzierska, E.; Kaczor, A.A. In Vitro and In Vivo Models for the Investigation of Potential Drugs Against Schizophrenia. Biomolecules 2020, 10, 160. [Google Scholar] [CrossRef]
- Forero, D.A.; González-Giraldo, Y. Integrative In Silico Analysis of Genome-Wide DNA Methylation Profiles in Schizophrenia. J. Mol. Neurosci. 2020, 70, 1887–1893. [Google Scholar] [CrossRef]
- De Rosa, A.; Fontana, A.; Nuzzo, T.; Garofalo, M.; Di Maio, A.; Punzo, D.; Copetti, M.; Bertolino, A.; Errico, F.; Rampino, A.; et al. Machine Learning algorithm unveils glutamatergic alterations in the post-mortem schizophrenia brain. Schizophr. Heidelb. 2022, 8, 8. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Frank, R.A.; Grant, S.G. Supramolecular organization of NMDA receptors and the postsynaptic density. Curr. Opin. Neurobiol. 2017, 45, 139–147. [Google Scholar] [CrossRef]
- Taylor, N.O.; Wei, M.T.; Stone, H.A.; Brangwynne, C.P. Quantifying Dynamics in Phase-Separated Condensates Using Fluorescence Recovery after Photobleaching. Biophys. J. 2019, 117, 1285–1300. [Google Scholar] [CrossRef]
- Serita, T.; Fukushima, H.; Kida, S. Constitutive activation of CREB in mice enhances temporal association learning and increases hippocampal CA1 neuronal spine density and complexity. Sci. Rep. 2017, 7, 42528. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Bonhoeffer, T.; Scheuss, V. Balance and stability of synaptic structures during synaptic plasticity. Neuron 2014, 82, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.P.; Mizusaki, B.E.; Sjöström, P.J.; van Rossum, M.C. Functional consequences of pre- and postsynaptic expression of synaptic plasticity. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2017, 372, e0153. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Manchia, M.; Marmo, F.; Vellucci, L.; Iasevoli, F.; Barone, A. Glycine Signaling in the Framework of Dopamine-Glutamate Interaction and Postsynaptic Density. Implications for Treatment-Resistant Schizophrenia. Front. Psychiatry 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Dell’Aversano, C.; Iasevoli, F.; de Bartolomeis, A. Homer splice variants modulation within cortico-subcortical regions by dopamine D2 antagonists, a partial agonist, and an indirect agonist: Implication for glutamatergic postsynaptic density in antipsychotics action. Neuroscience 2007, 150, 144–158. [Google Scholar] [CrossRef]
- Benjamin, K.J.M.; Chen, Q.; Jaffe, A.E.; Stolz, J.M.; Collado-Torres, L.; Huuki-Myers, L.A.; Burke, E.E.; Arora, R.; Feltrin, A.S.; Barbosa, A.R.; et al. Analysis of the caudate nucleus transcriptome in individuals with schizophrenia highlights effects of antipsychotics and new risk genes. Nat. Neurosci. 2022, 25, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Paget-Blanc, V.; Pfeffer, M.E.; Pronot, M.; Lapios, P.; Angelo, M.F.; Walle, R.; Cordelières, F.P.; Levet, F.; Claverol, S.; Lacomme, S.; et al. A synaptomic analysis reveals dopamine hub synapses in the mouse striatum. Nat. Commun. 2022, 13, 3102. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PSD Protein | Study Design | Methodology/ Samples | Species | Signaling | Functional Outcome | Authors |
|---|---|---|---|---|---|---|
| Arc | In vivo preclinical study | Golgi staining Western blot analysis | Mice (WT and SR−/−) | SR is the enzyme that converts L-serine to D- serine, a coagonist of NMDAR. SR−/− mice ↓ Arc in the hippocampus. | D-serine chronic treatment reverses the Arc expression alterations and rescues the hippocampal spine deficit in SR−/− mice. | Balu et al., 2014 [70] |
| DISC1 | Preclinical study | Immunoblot analysis Immunocytochemistry SDS-PAGE | Mice (KO, exon 3 deletion in LRRC7) | Densin, encoded by LRRC7, produces a high-affinity complex with α-CaMKII and α-actinin. Deletion of densin-180 ↓ mGluR5 and DISC1 levels. | Signaling defects by loss of densin alter the morphology of the spine and even produce behavioral endophenotypes that are SCZ- or ASD-like, | Carlisle et al., 2011 [71] |
| Preclinical study | Electrophysiology Immunoblotting HPLC | Rats (prenatal) | Ro61-8048 ↓ GluN2A and ↑GluN2B in embryos’ brains induce changes in sonic hedgehog at 24 h and ↑ neuronal excitability of coupled impulses. DISC1 was not modified. | The tryptophan–kynurenine pathway, influencing NMDAR, leads to synaptic changes in the embryonic and neonatal brain. | Forrest et al., 2013 [72] | |
| Preclinical study | UHVEM, TEM | Mice (DBZ KO) | In DBZ KO mice, ↓ dendritic arborization, ↑ density of spines (especially thin spines), and ↓ PSD number in the spines. | DBZ deficiency ↑ dendritic spine of cortical pyramidal neurons. | Koyama et al., 2015 [73] | |
| In vitro preclinical study | FRET, STORM Western blot Analysis | Mice | D2R and DISC1 complexes form clusters in the dendritic spines of striatal neurons. TAT-D2pep, assisted by NPY and GSK3β, ↓ D2R-DISC1 complexes, and even ↑ dendritic spines, synaptophysin, and PSD-95. | TAT-D2pep has a protective effect on neuronal morphology and dendritic spines. | Zheng et al., 2020 [74] | |
| Neuregulin | Preclinical study | Co-immunoprecipitation | Mice (erbB2/B4 KO and WT) | NRG1/erbB4 promotes dendrite growth in mature interneurons through kalirin. Phosphorylation in the C-terminal of kalirin-7 is important for the reduction in intraneuronal dendrite length. | The neural effects of kalirin (modulation of NRG1/erbB4 and dendrite growth) seem to be correlated with the SCZ onset. | Cahill et al., 2012 [75] |
| In vitro preclinical study | Immunostaining Immunoblot | Human lymphocytes (72 ASD patients) | NLGN at the level of PDZ3 interacted with PSD-95. Two patients with ASD had inherited missense mutations in conserved sites (T1033C/M152T and G1239T/V221L) of GPR85 related to G-protein interaction and signal transduction. Mutated GPR85 was preferentially accumulated, causing endoplasmic reticulum stress, and disrupting dendrite formation of hippocampal neurons, whereas, in GPR85 WT, this did not occur. | Mutated GPR85 in ASD patients disrupted dendrite formation through disconnection between GPR85 and NLGN via PSD-95, which is potentially implicated in ASD. | Fujita-Jimbo et al., 2015 [76] | |
| In vitro preclinical study | Immunocytochemistry Western Blot | Mice (ErbB4−/− and ErbB4+/+) | There was no difference in the size of the inhibitory synapse Gephyrin clusters in the dendrites of GABA-ergic cortical neurons between ErbB4−/− and ErbB4+/+ mice. The number of neuronal dendrites and Gephyrin inhibitory postsynaptic proteins in the ErbB4−/− group were lower. ErbB4−/− affected excitatory synapses in intermediate neuronal cells and inhibitory synapses in pyramidal neurons but did not result in changes in inhibitory ones. | The ErbB4 gene has no effect on either inhibitory synapses in interneurons or excitatory synapses in pyramidal neurons. NRG1 and ErbB4 signaling are necessary for the postnatal cortical GABA network. | Li et al., 2020 [77] | |
| Preclinical study | Semi-quantitativeRT-PCR Immunofluorescence | Mice | Neuronal ErbB-4 is most prevalent in GABAergic interneurons. The fraction of ErbB-4 receptors on the plasma membrane ↑ from 30% to 65% between 6 and 16 days in vitro; from stimulation by NRG, PSD-95 also ↑. In addition, TACE in hippocampal cultures is resistant to cleavage, especially the JM-b isoform. | At glutamatergic synapses, signaling is influenced by ErbB-4. | Logart et al., 2007 [78] | |
| Preclinical study | Western Blot Immunoprecipitation | Rats | After one day of being treated with MK-801, the rats exhibited ↑associations of ErbB4 with PSD-95 and NMDAR in the PFC, while only phosphorylated ErbB4 relative to ErbB4 was ↑in the hippocampal area, although these changes were detectable at 12 days. Moreover, such treatment led to a reduction in locomotor capacity and the onset of an anxiety-like phenotype. | Repeated blockade of NMDAR could modulate NRG1-ErbB4 signaling. | Li et al., 2013 [79] | |
| Preclinical study | Immunostaining Western Blot Electrophysiological | Mice | NRG1 ↑ PSD-95 and mEPSCs in GABAergic interneurons, while it had no effects on excitatory synapses in glutamatergic neurons. In addition, the deletion of ErbB4 in parvalbumin-positive interneurons results in reduced mEPSCs. | ErbB4 has a pivotal role in the synaptogenesis of excitatory synapses, while NRG1 has a specific effect on synapses of GABAergic interneurons. | Ting et al., 2011 [80] | |
| PSD-95 | Preclinical study | Western blot analysis | Mice (GlyT1+/− and SR−/−) | SR−/− mice had elevated NR1 and NR2A NMDAR protein levels in the hippocampus but not in PFC as in GlyT1+/− mice. In contrast, there were no changes for AMPAR (GluR1, GluR2) or PSD95 in either region. GlyT1+/− mice had high expression of NR1, NR2A, GluR1 and GluR2 in PSD. | Genetic perturbation and ionotropic glutamate receptor alterations in the PSD of hippocampal neurons are implicated in SCZ. | Balu et al., 2011 [81] |
| Post-mortem study | Immunoprecipitation Western blot analysis | Mice and human brain tissue | NMDA receptor complexes in the PSD are ↑ in the SCZ. The DLPFC of SCZ cases shows an ↑ in PSD-95 and erbB4 and an ↓ in RPTPa and dysbindin-1, each of which reduces Src activity through protein interaction with Src. | In SCZ, there is a reduction in NMDA receptor signaling at the post-receptor level, and Src is probably responsible for these dysregulations. | Banerjee et al., 2015 [82] | |
| Preclinical study | Immunoprecipitation Western blot analysis | Mice (Dlgap1 WT, HT, and KO) | DLGAP1 protein is localized in glutamatergic neurons and is a component of protein complexes organized by PSD95. | Dlgap1 KO mice had sociability deficits and disruption of protein interactions in PSD. | Coba et al., 2018 [83] | |
| Preclinical study | Co-immunoprecipitation Western blot | Mice (PSD-95−/−) | During adolescence in the mice analyzed, there was an ↑ in total protein levels of the NMDAR subunits GluN1 and GluN2B, with an ↓ in the AMPAR subunit GluA1. | PSD-95−/− mice showed a lack of sociability and deficits in learning and working memory with altered mPFC synaptic function. | Coley et al., 2019 [84] | |
| Preclinical study | Immunocytochemistry Immunoprecipitation Electrophysiology | Mice | SULT4A1 modulates neuronal branching and dendritic spine formation; also, by negatively regulating the catalytic activity of Pin1 for PSD-95, it facilitates the expression and synaptic function of NMDAR. | Inhibition of Pin1 reverses the pathological phenotypes restoring dendritic spine density and improving NMDAR-mediated synaptic transmission. | Culotta et al., 2020 [85] | |
| Preclinical study | Immunoprecipitation Mass spectrometry | Mice (PSD-95TAP/+) | Homozygous mice showed no alterations in PSD-95 expression, subcellular localization, and synaptic function. | PSD-95 is a SCZ susceptibility protein. | Fernandez et al., 2009 [86] | |
| Preclinical study | Electrophysiology Immunoblotting | Rats | Ro61-8048 administration determined sonic hedgehog expression ↑, as did levels of doublecortin and PCNA, suggesting activation of neurogenesis. Moreover, the PSD-95 ↑. | The kynurenine pathway determined functional synaptic changes in the embryonic and neonatal nervous system influencing the expression of proteins that modulate NMDAR. | Forrest et al., 2013 [72] | |
| In vivo preclinical study | Co-immunoprecipitation Immunocytochemistry Immunohistochemistry Electrophysiology | Mice (SNAP-25 WT and HT) | SNAP-25 includes PSD-95 and p140Cap. Acute downregulation of SNAP-25 in CA1 affects the number of dendritic spines, and hippocampal neurons from mice heterozygous for SNAP-25 show reduced densities of spines and alterations in PSD-95. | SNAP-25 is important in controlling PSD-95 and may contribute to SCZ through an effect on postsynaptic function and plasticity. | Fossati et al., 2015 [87] | |
| In vitro preclinical study | Immunostaining Immunoblot | Human lymphocytes (72 ASD patients) | The C-terminal sequence of GPR85 at the level of PDZ1 interacted with PSD-95, while NLGN at the level of PDZ3 interacted with PSD-95. | GPR85 carrying the mutations disturbed dendrite formation that could be implicated in the pathogenesis of ASD through the associated NLGN-PSD-95 receptor complex. | Fujita-Jimbo et al., 2015 [76] | |
| Preclinical study | Co-immunoprecipitation Immunohistochemistry Western Blot | Rats | The expression of PSD-95 ↑ in the medial PFC of maternal separation-exposed adolescents, while NR2A did not significantly ↑. This effect resulted in behavioral alterations. | The ↑ of NR2A in PFC is related to SCZ and acts largely through its association with the postsynaptic protein PSD-95. | Ganguly et al., 2015 [88] | |
| Preclinical study | BiFC Co-immunoprecipitation Immunoblotting Immunocytochemistry | Rats | Calcyon interacting with PSD-95 is then able to form a ternary complex with D1DR. Calcyon when phosphorylated ↑ the association with PSD-95. | Calcyon modulates trafficking of D1DR by forming a ternary complex with it via PSD-95. | Ha et al., 2012 [89] | |
| Preclinical study | RT-PCR cDNA Constructs | Mice (Shank3- overexpressing TG) | The expression of Gpr85 was ↑ in multiple brain regions of the mice, while the mRNA levels of Gpr27 and Gpr173 ↓. | Gpr85 was colocalized with PSD-95 and Shank3 mice brain. | Jin et al., 2018 [90] | |
| Preclinical study | Immunoprecipitation Western bot | Rats | One day after MK-801 administration in rats, the associations of ErbB4 with PSD-95 and NMDAR were ↑ in the PFC, whereas in the hippocampus only phosphorylated ErbB4 ↑. These effects tended to fade chronically, furthermore, long-term treatment resulted in reduced locomotor capacity and the onset of an anxiety-like phenotype. | NRG1-ErbB4 signaling could be modulated by repeated blockade of NMDAR with PSD-95. | Li et al., 2013 [79] | |
| In vitro preclinical study | Immunocytochemistry Western blot | Mice (ErbB4−/− and ErbB4+/+) | PSD-95 on the neuronal surface was significantly lower than in 1NMPP1, while Gephyrin on the neuronal surface was equally present. | The density of PSD-95 in GABAergic neurons was not different in the controls. | Li et al., 2020 [77] | |
| In vivo preclinical study | Immunocytochemistry | Mice | Slitrk2 directly interacts with PSD-95 through a SH3 domain binding motif. Furthermore, PSD-95 induces robust clustering of Slitrk2 in 293T cells. The deletion of the SH3 domain in PSD-95 or the SH3 domain binding motif in Slitrk2 reduces this clustering. | PSD-93 and PSD-95 can form molecular complexes with NRG and K+ channel and are associate with Slitrk2 in postnatal mouse brain. | Loomis et al., 2020 [91] | |
| Preclinical study | Immunocytochemistry Immunoprecipitation Western bot | Mice | SR binds both PSD-95 and stargazin, this results in inhibition of the SR enzyme. This complex is blocked, triggering the synthesis of D-serine, by activation of AMPA receptors. | The interactions of SR-stargazin-PSD-95 modulated NMDA/AMPA receptor. | Ma et al., 2014 [92] | |
| Preclinical study | Western bot ELISA | Rats | The density of dendritic spines and their content of PSD-95, Syn, AMPA receptors, and BDNF ↓ in AI-injured rats. In addition, neural architecture was changed with thin, mushroom-shaped, stubby and broad spines. | In AI-injured animals, working memory and social behavior were altered, as well as PSD-95 and AMPA receptors. | Martinez-Torres et al., 2021 [93] | |
| Preclinical study | Co-immunoprecipitation Electrophysiology Western bot | Mice (PSD-95+/−) | In HT mice, there was an ↑ in α1 subunit of the GABAA receptor and an ↑ in sIPSCs in mPFC pyramidal neurons, with a significant ↑ in evoked IPSCs, leading to excitatory–inhibitory dysregulation in such mice. PSD-95 deficiency enhances the functioning of inhibitory synapses through upregulation and trafficking of NLGN2 and reduction of GSK3β activity. | PSD-95 deficiency results in effects on GABAergic transmission in mPFC. | McEachern et al., 2020 [94] | |
| Preclinical study | Mice (HB-EGF KO) | KO mice had behavioral abnormalities ameliorable by antipsychotic treatment, altered dopamine and serotonin levels, ↓ spines in PFC neurons, ↓ CaMK II, NR1 and PSD-95. | Alterations in HB-EGF signaling may predispose SCZ, through dysregulation, of many factors, including PSD-95. | Oyagi et al., 2009 [95] | ||
| SAP97 | Preclinical study | Western Blot | Mice | CRFR1 acts with SAP97, HEK 293, and cortical brain lysates. This interaction depends on an intact PDZ binding motif at the carboxyl-terminal tail end of CRFR1. SAP97 is recruited to the plasma membrane in HEK 293 cells expressing CRFR1, and mutation of the PDZ-binding motif of CRFR1 results in the redistribution of SAP97 in the cytoplasm. SAP97 overexpression inhibits internalization of CRFR1. Knockdown with shRNA of SAP97 did not affect cAMP, but knockdown of SAP97 attenuated CRFR1-stimulated phosphorylation of ERK1/2. | SAP97 interactions with CRFR1 attenuate CRFR1 endocytosis and SAP97 is involved in the coupling of G-protein-coupled receptors to ERK1/2 signaling pathway activation. | Dunn et al., 2013 [96] |
| Preclinical study | qPCR Western Blot RNA sequencing | Mice (SAP97-cKO) | RNA sequencing of hippocampi from male control and SAP97-cKO animals identified 67 transcripts regulated by SAP97. Male-specific cognitive impairment and female-specific motor impairment were found in SAP97-cKO mice, while other behaviors remained essentially unchanged. | Loss of SAP97 is associated with SCZ, and it may have a contribution in the behavior. | Gupta et al., 2018 [97] | |
| Shank | Preclinical study | Immunocytochemistry Immunohistochemistry RT-PCR Western Blot MicroArray Mass-spectrometry | Mice | De novo mutation R1117X in ProSAP2/Shank3 leads to its accumulation in the nucleus. This alters the transcription of some genes involved in SCZ, such as Synaptotagmin 1 and LRRTM1. In addition, hippocampal neurons with the ProSAP2/Shank3 SCZ mutation show altered ratio and reduced dendritic branching. | The uncoupling of nuclear shuttling of ProSAP2/Shank3 from synaptic activity may represent one of the mechanisms underlying the pathogenesis of SCZ. | Grabrucker et al., 2014 [61] |
| Preclinical study | Chromatography NMR spectroscopy | Mammalian cells | Mammalian Shank proteins have identical and typical SH3 folding motifs but unusual target binding pockets. | There is an atypical interaction where SH3 domains binds to a region of Cav1.3. | Ishida et al., 2018 [98] | |
| Preclinical study | RT-PCR cDNA Constructs | Mice (Shank3- overexpressing TG) | In Shank3 TG mice several myelin-related genes were downregulated specifically in mPFC, but not in the striatum or hippocampus. mRNA levels of Gpr85 ↑ in multiple brain regions of the mice brain, while mRNA levels of Gpr27 and Gpr173 ↓ in cortex and striatum. | Molecular changes of overexpressing Shank3 in mice brains could be implicated in the pathogenesis of SCZ. | Jin et al., 2018 [90] | |
| Preclinical study | Liquid chromatography–tandem MS | Mice | LTP results in the reorganization of PSD, which in turn links glutamate receptor signaling to kinases and phosphatases as well as target proteins modulated by protein phosphorylation. | Phosphoproteins regulated by LTP, including the scaffold proteins Shank3, Syngap1, Dlgap1 and Dlg4, were a risk factor for SCZ and autism spectrum disorder. | Li et al., 2016 [99] | |
| Preclinical study | Biochemistry Mass spectroscopy | Mice | There is a central PSD scaffold at age 14, but while the NMDAR subunits are already linked to MAGUKs and DLGAPs protein complexes, the downstream components are held in separate protein complexes organized by SHANKs. There is a continuous expansion of functional groups and ↑ associations to glutamate receptors in the upper layers and proteins with embedded enzyme activities in the middle and lower scaffold layers. The scaffold interactors form a more heterogeneous set of protein complexes than the basic PSD scaffolds. | The network of protein interactions of the adult PSD is connected through a core scaffold component that uses specific hubs for localization. Instead, a number of PSD scaffold interactors may also associate in protein networks enriched in “housekeeping” protein interactions early in development that are not exclusive to neurons. | Li et al., 2017 [100] | |
| Preclinical study | Chromatography | Rats | The binding specificities of PDZ domains of different Shank are similar. The structure in solution of Shank3 in tandem containing the SH3 and PDZ domains showed that the two domains are close to each other. The SH3 domain did not affect the affinity of the PDZ domain toward short target peptides. | The R536W mutation of Shank3 (implicated in SCZ) in the binding between the domains had no effect on the structure or peptide interactions of the Shank3 SH3-PDZ unit. | Ponna et al., 2018 [101] |
| PSD Protein | Study Design | Methodology/ Samples | Species | Drug Intervention | Functional Outcome | Authors |
|---|---|---|---|---|---|---|
| Arc | Preclinical study | In situ hybridization | Rats | HAL (In acute) | HAL ↑Arc in NAc, dlCP, dmCP, vmCP and vlCP. MIN ↓Arc in the same regions. Moreover, HAL add-on to MIN ↓ Arc in ACC, MAC, M1, SS, and IC. | Buonaguro et al., 2017 [106] |
| Preclinical study | In situ hybridization, Probe radiolabeling | Rats | ASE, HAL, OLA (In acute) | HAL ↑ Arc in NAc (core and shell), dmCP, dlCP, and vlCP; and HAL ↓ Arc in ACC, M2, SS, and IC. ASE ↑ Arc in NAc core, dlCP, and vlCP. ASE or OLA ↓ Arc in ACC, M1, M2, dlCP, vlCP, and NAc core. | de Bartolomeis et al., 2015 [107] | |
| Preclinical study | In situ hybridization, Western blot analysis | Rats | HAL (In acute) | HAL ↑ Arc expression in dlCP, dmCP, and vlCP and ↓ Arc in M2, SS, and IC. | de Bartolomeis et al., 2018 [108] | |
| Preclinical study | In situ hybridization, Probe radiolabeling | Rats | HAL, SER (In acute) | Arc was ↑ by HAL in dlCP, dmCP, vmCP, vlCP, and NAc, (only core). SER has no significant effects on Arc expression. | Iasevoli et al., 2010 [109] | |
| DISC1 | Post-mortem study | Immunohistochemistry, Immunolabeling, Western blot analysis | Human (7 normal brain tissue) | - | The presence of DISC1 in multiple types of frontal and parietal cortex (BA 4, 9, 39, and 46) synapses, in ribosomes, dendritic trees, microtubules, Golgi apparatus, and multivesicular bodies suggests that DISC1 may participate in synaptic activity. | Kirkpatrick et al., 2006 [110] |
| Homer | In vitro preclinical study | Immunocytochemistry, Immunoblotting, Semiconductor sequencing, PCR | Human (98 patients with SCZ history and 484 controls) | - | Eighteen genes were sequenced for PSD-related proteins (NLGN, SHANK, DLGAP Homer) in SCZ patients with a psychiatric family history. In the SCZ patients’ group, a mutation of HOMER2 (c.826A>G, rs373795200) was found. | Hu et al., 2020 [10] |
| Post-mortem study | Immunoblotting | Human (20 SCZ patients and 20 controls) | - | Alterations of the scaffold protein Homer1 (+42.9%) and Homer1b/c (− 24.6%) were evidenced in the CA1 hippocampal region in SCZ patients. | Matosin et al., 2016 [111] | |
| Preclinical study | In situ hybridization | Rats | HAL | HAL ↑ Homer1a in dmCP, dlCP, vmCP, and vlCP. On the contrary, Homer1b/c expression was not variated. | Buonauguro et al., 2017 [106] | |
| Preclinical study | In situ hybridization | Rats | HAL, ASE, OLA | HAL shifted the ratio toward Homer1b in ACC, M2, M1, and IC, while shifted the ratio toward Homer1a in the dlCP and NAc. ASE ↑ Homer1b in the IC, and SS and ↑ Homer1a in dlCP. OLA ↑ Homer1a in dlCP. | Buonauguro et al., 2017 [112] | |
| Preclinical study | In situ hybridization | Rats | HAL, OLA | Both AT (HAL and OLA) ↑ Homer expression in the NAc. | de Bartolomeis et al., 2002 [113] | |
| In vivo preclinical study | In situ hybridization, Radiolabeling, Histochemistry | Rats | Lithium, VPA (In chronic) | Lithium and VPA ↓ Homer1b/c in M1, M2, IC, dmCP, vmCP, and dlCP. Moreover, VPA ↓ Homer1b/c in NAc core. | de Bartolomeis et al., 2012 [104] | |
| Preclinical study | In situ hybridization | Rats | KET, MEM, MK-801 | KET ↑Homer1a in IC, and ↓Homer1b in M1, dlCP, and MAC. MEM ↑Homer1b in the MAC. KET and MK-801 ↑ Homer1a/Homer1b ratio, while MEM ↓ it. | de Bartolomeis et al., 2013 [114] | |
| Preclinical study | In situ hybridization | Rats | ASE, HAL, OLA | OLA ↑ Homer1a in dmCP, dlCP, vlCP, vmCP, NAc shell, AC, M2, M1, and IC. HAL ↑ Homer1a in dmCP, dlCP, vlCP, vmCP, and NAc, in addition ↑ Homer1b/c in M1. ASE ↑ Homer1a in AC, M2, M1, SS, dmCP, dlCP, vlCP, vmCP, and ↑ it and Homer1b/c in NAc core. All AT ↓ Homer1b/c in AC, M2 and M1. | de Bartolomeis et al., 2015 [107] | |
| Preclinical study | In situ hybridization | Mice (Ddo−/−and Ddo+/+) | PCP (in acute) | Ddo−/− naive mice exhibited ↓ Homer1a in the PFC, PrL, VO, LO, and DLO, ↑ Homer1b/c in the striatum. PCP ↑ Homer1a in the PFC of Ddo−/− mice. | de Bartolomeis et al., 2015 [115] | |
| Preclinical study | In situ hybridization | Rats | AMS, HAL | AMS ↑ Homer1a in the core of NAc, while HAL ↑ it in the VL and M1. Both drugs ↑ Homer1a in dmCP and vmCP. Homer1b/c in the striatum was more ↑ by AMS compared to HAL. | de Bartolomeis et al., 2016 [116] | |
| Preclinical study | In situ hybridization, Western blot | Rats | HAL | Homer1a was ↑ by HAL in vmCP, dmCP, and NAc core. | de Bartolomeis et al., 2018 [108] | |
| Preclinical study | In situ hybridization, Histochemistry | Rats | KET | Homer1a was ↑ by KET in the vmCP and NAc. KET ↑ α-CaMKII in dlCP, vmCP, dmCP, and NAc shell, while β-CaMKII was not affected. DAT ↑ in VTA and SNc. | Iasevoli et al., 2007 [117] | |
| Preclinical study | Radiolabeling | Rats | HAL, TER, Others (D1/D2/D3/D4 antagonists) | Homer1a was ↑ in NAc by HAL and in vlCP by TER. In MAC and M1, it was ↑ by D1, D2 and D3 antagonists. Homer1b was ↓ by TER. In the vlCP, vmCP, and NAc shell. Furthermore, HAL ↓Homer1b in vlCP. | Iasevoli et al., 2009 [118] | |
| Preclinical study | In situ hybridization, Radiolabeling | Rats | HAL, SER (In acute and chronic) | In acute: HAL but not SER, ↑Homer1a in dlCP, vlCP, vmCP, and dmCP. In addition, ↓ Homer1a in ACC, MAC, and M1. Both drugs ↓ Homer1a in SS and IC. In chronic: Homer1b/c was ↑ by HAL and SER in ACC, MAC, M1, SS, and IC. | Iasevoli et al., 2010 [109] | |
| Preclinical study | Autoradiography, In situ hybridization | Rats | CLO, HAL, ZIP (In acute and chronic) | In acute: HAL, ZIP, and CLO ↑ Homer1a in dmCP, dlCP, vmCP, vlCP, and NAc. CLO and ZIP ↑ Homer1a in ACC, SS, IC, MAC (only CLO), and M1 (only ZIP). In chronic: HAL ↑ Homer1a in dmCP, dlCP, vmCP, vlCP, and NAc; ZIP only in dlCP, vlCP, and core of NAc. ZIP ↑ Homer1b in dlCP. | Iasevoli et al., 2011 [119] | |
| Preclinical study | In situ hybridization | Rats | HAL, MEM (In acute) | Homer1a was ↑ by HAL and MEM in dlCP, vlCP, vmCP, and dmCP. MEM significantly induced Homer1b/c variation in dmCP, vmCP, M1, SS, and IC. | Iasevoli et al., 2014 [120] | |
| Preclinical study | In situ hybridization | Rats | ASE, HAL, OLA (In acute and chronic) | HAL in acute ↑ Homer1a in dlCP, vlCP, vmCP, dmCP, and core of NAc more than OLA. No effects on Homer were found by ASE. Compared with chronic administration, acute AT administration had a greater effect on Homer1a expression. | Iasevoli et al., 2020 [121] | |
| Neuregulin | In vitro preclinical study | Immunocytochemistry, Immunoblotting, Semiconductor sequencing, PCR | Human (98 SCZ patients with family history, 484 nonrelated with SCZ, 533 controls) | - | PSD-related genes, such as NLGN, SHANK, and DLGAP, have rare functional mutations (e.g., NLGN3 c.584G>A p.R195Q rs190164205) that could be related to pathogenesis of SCZ. | Hu et al., 2020 [10] |
| Post-mortem study | Immunoprecipitation, Immunoblotting | Human brain tissue (14 SCZ patients, 14 controls | - | NRG1 and erbB4 levels in PFC did not differ between groups. SCZ patients showed ↑ erbB4-PSD-95 interactions. In addition, stimulation of NRG1 downregulates NMDA receptor activation in the PFC. | Hahn et al., 2006 [122] | |
| Post-mortem study | Immunoprecipitatiom, Immunoblotting | Human PFC (14 SCZ patients, 14 controls) | - | NMDA in SCZ patients appeared sensitive to the NRG1inhibition, that mediates the stimulation of ErbB4. The Erb-PSD-95-NMDA complex is implicated in SCZ. | Morrison et al., 2007 [123] | |
| Preclinical study | ECL, Western blot | Rats (Perinatal model) | PCP | PCP treatment in the cortex determined ↓NRG1 and ErbB4 at PN12 and ↑in ErbB4 and p-ErbB4 at 5 weeks, and ↓ErbB4 and p-ErbB4 together with an ↑Nrg1 at 20 weeks. In contrast, in the hippocampus, only determined ↓ p-ErbB4 at 20 weeks. | Du Bois et al., 2012 [124] | |
| PSD-95 | Post-mortem study | Real-time quantitative, RT-PCR | Human brain tissue (1539 SCZ patients, 2012 controls) | - | PSD95 group (DLG 1-4) is the most important postsynaptic density component in glutamatergic synapses. SCZ patients’ haplotypes of DLG4, rs2242449, rs17203281, rs390200, rs222853, and rs222837 had A-C-C-A/G-C-C-A accumulated sequences. | Balan et al., 2013 [125] |
| Post-mortem study | Western blot analysis | Human brain tissue (37 SCZ patients, 37 controls) | - | Patients with SCZ had 30% ↓ PSD-95 and 20% ↓ NR1 proteins, so there were fewer functional NMDA receptors in postsynaptic density. | Catts et al., 2015 [126] | |
| Post-mortem study | Western blot analysis | Human brain tissue (SCZ patients: 36ACC + 35DLPFC and controls: 33ACC + 31DLPFC) | - | In ACC, pS295, PSD-95, Rap2, JNK1, and JNK2 were ↓. In the DLPFC, the expression of Cdk5, Rack1, pS295 PSD-95, and NR2B were ↑. | Funk et al., 2012 [127] | |
| Post-mortem study | Western blot analysis, TaqMan PCR | Human brain tissue, mice, rats | HAL (rats) | In SCZ group, there was ↓ PSD-95. The beta variant of PSD-95 was ↑ in the GluN1 KD mouse model of SCZ, while the alpha variant was ↑ in rats HAL-treated. | Funk et al., 2017 [128] | |
| Post-mortem study | TaqMan PCR | Human brain tissue (259 SCZ patients, 188 controls) | - | Six SNPs (rs314253 C/T, rs13331 A/G, rs2242449 C/T, rs390200 A/G, rs507506 A/G, rs739669 A/G) of PSD-95 seemed to not represent a risk factor for SCZ. | Kawashima et al., 2006 [129] | |
| Association study | Maldi-Tof mass, spectrometry | Human (248 SCZ patients, 208 controls) | - | Two PSD-95 variants (rs2521985 and rs17203281) were not associated with SCZ. | Tsai et al., 2007 [130] | |
| Association study | PCR, Sanger sequencing | Human (1685 SCZ patients in total, 574 ASD in total, 1793 controls) | - | A carrier of the DLG1-G344R PSD-95 variant was identified in the SCZ group. The DLGAP2-R604C variant was present in ASD group. | Xing et al., 2016 [131] | |
| Post-mortem study | Western blot analysis | Human brain (13 SCZ patients, 8 controls) | - | NR2B and PSD-95 expression in the dorsolateral PFC in the SCZ was ↓. | Kristiansen et al., 2010 [132] | |
| Post-mortem study | In situ Hybridization, Western blotting | Human brain tissue (24 SCZ patients, 16 controls) | - | PSD-95 expression was ↓ in the ACC. | Kristiansen et al., 2006 [133] | |
| Post-mortem study | Immunoprecipitation, TEM, Western blot | Human frontal cortex (15 for each group: SCZ, MDD, BPD, controls) | - | NR2A, PSD-95, CaMKIIα, and CaMKIIβ was ↓ in SCZ. While only NR2A was ↓ in MDD. | Matas et al., 2021 [134] | |
| Post-mortem study | Immunoblotting | Human (20 SCZ patients and 20 controls) | - | Patients in the CA1 region presented ↓ PSD95, synaptophysin, and mGluR1. | Matosin et al., 2016 [111] | |
| Post-mortem study | In situ Hybridization | Human brain tissue (16 SCZ patients, 7 controls) | - | NF-L, NR1, GluR5, and PSD-95 were ↑, while SAP102, PSD-93. and yotiao were ↓ in SCZ. | Mueller et al., 2004 [111] | |
| Preclinical study | Immunohistochemistry, Western blot | Mice and human brain tissue | - | Synaptic dysbindin-1 was ↓ in the SCZ; synaptophysin or PSD-95 levels did not change. | Talbot et al., 2011 [135] | |
| Post-mortem study | Immunoautoradiography, Western blot | Human brain tissue (15 for each group: SCZ, MDD, BPD, controls) | - | Reduction of PSD-95 at molecular layer glutamate synapses can alter connections to other hippocampal regions. | Toro et al., 2005 [136] | |
| Preclinical study | In situ hybridization | Rats | HAL, OLA | PSD-95 expression was not changed by administration of typical or atypical AT. | de Bartolomeis et al., 2002 [113] | |
| Preclinical study | In situ hybridization | Rats | KET, MEM, MK-801 | KET and MK-801 ↓PSD-95 in dmCP and dlCP. MEM ↑PSD-95 in dlCP and vmCP. | de Bartolomeis et al., 2013 [114] | |
| Preclinical study | In situ hybridization | Mice (Ddo−/− and Ddo+/+) | PCP | Ddo−/− naive mice present ↓PSD-95 in the striatum and cortex. In M1, mRNA expression of PSD-95 was higher in Ddo−/− PCP mice than in Ddo−/− mice. In addition, gene expression was higher in Ddo+/+ PCP mice than in KO mice in both M1 and PrL. | de Bartolomeis et al., 2015 [115] | |
| Preclinical study | In situ hybridization | Rats | ASE, HAL, OLA | In the cortex, striatum, and NAc, PSD-95 expression was ↑ by both ASE and OLA. While PSD95 was ↑ by HAL in AC, M1, M2, I, dmCP, dlCP, vmCP, NAc. | de Bartolomeis et al., 2015 [107] | |
| Preclinical study | Autoradiography, In situ hybridization | Rats | CLO, HAL, ZIP | PSD-95 ↑ in the striatum and ACC by administration of HAL, and in the striatum and cortex (ACC, MAC and M1) by ZIP. | Iasevoli et al., 2011 [119] | |
| Preclinical study | Co-mmunoprecipitation, Western blot analysis | Mice | D-serine | Application of D-serine ↑ SR interactions with PSD-95 and NR1 and ↑ the number of glutamatergic synapses positive at VGLUT1 and PSD-95. D-serine/SR are associated with PSD-95 and NMDAR in postsynaptic neurons and correlate with the stability of glutamatergic synapses during synaptogenesis. | Lin et al., 2016 [137] | |
| Preclinical study | Electrophysiology, Densitometry, Western blot analysis | Mice (Ns−/−) | - | Ns-/- mice showed cognitive deficits and ↓ spinal synapse density in CA1 of the hippocampus, while PSD-95 ↑ in Ns−/− mice. These mice also showed a ↓ LTP. | Reumann et al., 2017 [138] | |
| In vitro preclinical study | Immunofluorescence, Western blot analysis | Rats | APZ, CLO HAL | APZ and CLO ↑ PSD95 protein levels, the number of spines, phosphorylated Akt Thr308 and Ser473, and phosphorylated GSK-3 beta Ser9. In contrast, HAL or an inappropriate concentration of CLO ↓ them. | Takaki et al., 2018 [139] | |
| Preclinical study | Immunohistochemistry Transmission electron microscopy assay Western blot analysis | Rats | KET | Ketamine resulted in ↓ BDNF and PSD-95 levels in PCC. Vinpocetine can reverse synaptic ultrastructure by upregulating BDNF-bound PSD-95 reducing ketamine-induced SCZ-like effects in rats. | Xu et al., 2019 [140] | |
| Preclinical study | PCR Amplification Direct PCR-Sequencing Reaction | Human (523 SCZ patients, 596 controls) | - | Increased expression of the DLGAP2 gene is implicated in the pathogenesis of SCZ. | Li et al., 2014 [141] | |
| SAP97 | -Post-mortem study -Preclinical study | Western blot analysis | -Human brain (34 SCZ patients, 41 controls) -Rats | HAL (rats) | SAP97 levels is alterated in the PFC of SCZ patients, and this modification of PDZ protein expression is not correlated to AT used. | Toyooka et al., 2002 [142] |
| SAP102 | Post-mortem study | In situ Hybridization, Histochemistry | Human brain tissue (60 subjects) | - | In the SCZ, MDD and BD NR1 is ↓. There is a ↓ in NR2A in the SCZ and MDD and a ↓ in NR2C in the SCZ. The expression of SAP102 was ↓ in BD. | Beneyto et al., 2008 [143] |
| Post-mortem study | Western blotting | Human brain tissue (15 SCZ patients, 8 controls) | - | NR2B and PSD95 are ↑ in the dorsomedial thalamus but not in the ventral thalamus of SCZ patients. While NF-L and SAP102 levels are not different between groups. | Clinton et al., 2006 [144] | |
| Post-mortem study | In situ Hybridization | Human striatum tissue (60 subjects overall, 3 gropus 15 SCZ/ BD /MDD) | - | PSD-95 and SAP-102 in BD and SAP-102 in MDD and SCZ are ↓ in striatum. | Kristansen et al., 2005 [145] | |
| Post-mortem study | In situ Hybridization Western blotting | Human brain tissue (24 SCZ patients, 16 controls | - | A significant ↑ of NR1C20 in the ACC was found in the SCZ patients. Furthermore, the expression of NF-L in DLPFC and PSD-95 and PSD-93 in the ACC are changed. SAP102 expression do not have any modifications. | Kristansien et al., 2006 [133] | |
| Post-mortem study | In situ Hybridization | Human brain (24 subjects overall, 3 gropus 8 SCZ/ BD /MDD) | - | In BD, not in SCZ, ↓ NR1 and NR2A and SAP102 in hippocampus (CA1, CA2, CA3, CA4, DG, and Sub). | McCullmsmith et al., 2007 [146] | |
| Post-mortem study | In situ Hybridization | Human brain (16 SCZ patients, 7 controls) | - | SAP102 was poorly expressed in the SNc | Mueller et al., 2004 [147] | |
| Shank | Post-mortem study | Western blotting dot blots | Human brain tissue (20 SCZ patients and 20 controls) | - | Alterations of proteins involved in CME (such as Dynamin-1, adaptor protein 2) and proteins interacting with NMDA were related to SCZ. PSD, NMDA-interacting proteins and endocytosis-related proteins contribute to the pathophysiology of SCZ. | Föcking at al., 2015 [69] |
| In vitro preclinical study | Immunocytochemistry, Immunoblotting, Semiconductor sequencing, PCR | Human (98 SCZ and 484 controls) | - | PSD-related genes (such as NLGN, SHANK and DLGAP) had rare functional mutations that could alter protein expression in some patients with SCZ. | Hu et al., 2020 [10] | |
| Preclinical study | qRT-PCR Immunocytochemistry Western blotting | Human (Patients with 22q13 deletion) | - | PMDS presented ↓ of Shank3 expression in neurons. | Shcheglovitov et al., 2013 [148] | |
| In vivo preclinical study | In situ hybridization, Histochemistry, Radiolabeling | Rats | Lithium, VPA (in chronic) | Both mood stabilizers ↓ expression of Shank, IP3R, and Homer1b/c correspondingly in cortex and dorsolateral caudate-putamen, whereas only VPA reduced gene expression in all other striatal subregions. | De Bartolomeis et al., 2012 [104] | |
| Spinophilin | Post-mortem study | Western blot analysis | Human brain (24 subjects SCZ: 12 AT-free, 12 AT-treated; and 24 controls) | - | Spinophilin lower band (80–95 kDa) showed a significant ↓ in AT-treated dorsolateral PFC of SCZ subjects compared to controls (but not in AT-free). | Brocos et al., 2021 [149] |
| Post-mortem study | qRT-PCR analysis | Human brain tissue | - | There was a significant negative correlation with interneuronal markers, parvalbumin and somatostatin, while a positive correlation with spinophilin, but not with PSD-95. | Catts et al., 2012 [150] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bartolomeis, A.; Vellucci, L.; De Simone, G.; Mazza, B.; Barone, A.; Ciccarelli, M. Dysregulated Signaling at Postsynaptic Density: A Systematic Review and Translational Appraisal for the Pathophysiology, Clinics, and Antipsychotics’ Treatment of Schizophrenia. Cells 2023, 12, 574. https://doi.org/10.3390/cells12040574
de Bartolomeis A, Vellucci L, De Simone G, Mazza B, Barone A, Ciccarelli M. Dysregulated Signaling at Postsynaptic Density: A Systematic Review and Translational Appraisal for the Pathophysiology, Clinics, and Antipsychotics’ Treatment of Schizophrenia. Cells. 2023; 12(4):574. https://doi.org/10.3390/cells12040574
Chicago/Turabian Stylede Bartolomeis, Andrea, Licia Vellucci, Giuseppe De Simone, Benedetta Mazza, Annarita Barone, and Mariateresa Ciccarelli. 2023. "Dysregulated Signaling at Postsynaptic Density: A Systematic Review and Translational Appraisal for the Pathophysiology, Clinics, and Antipsychotics’ Treatment of Schizophrenia" Cells 12, no. 4: 574. https://doi.org/10.3390/cells12040574