
Role of Cholesterol 25-Hydroxylase (Ch25h) in Mediating Innate Immune Responses to Streptococcus pneumoniae Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Streptococcus pneumoniae Infection
2.3. Cell Isolation and Culture
2.4. Bacterial Labeling
2.5. In Vivo Procedures and Tissue Collection
2.6. RNA Purification and Real-Time PCR
2.7. Cytokine Quantification in Lung Tissue
2.8. Transmission Electron Microscopy (TEM)
2.9. Statistical Analysis
3. Results
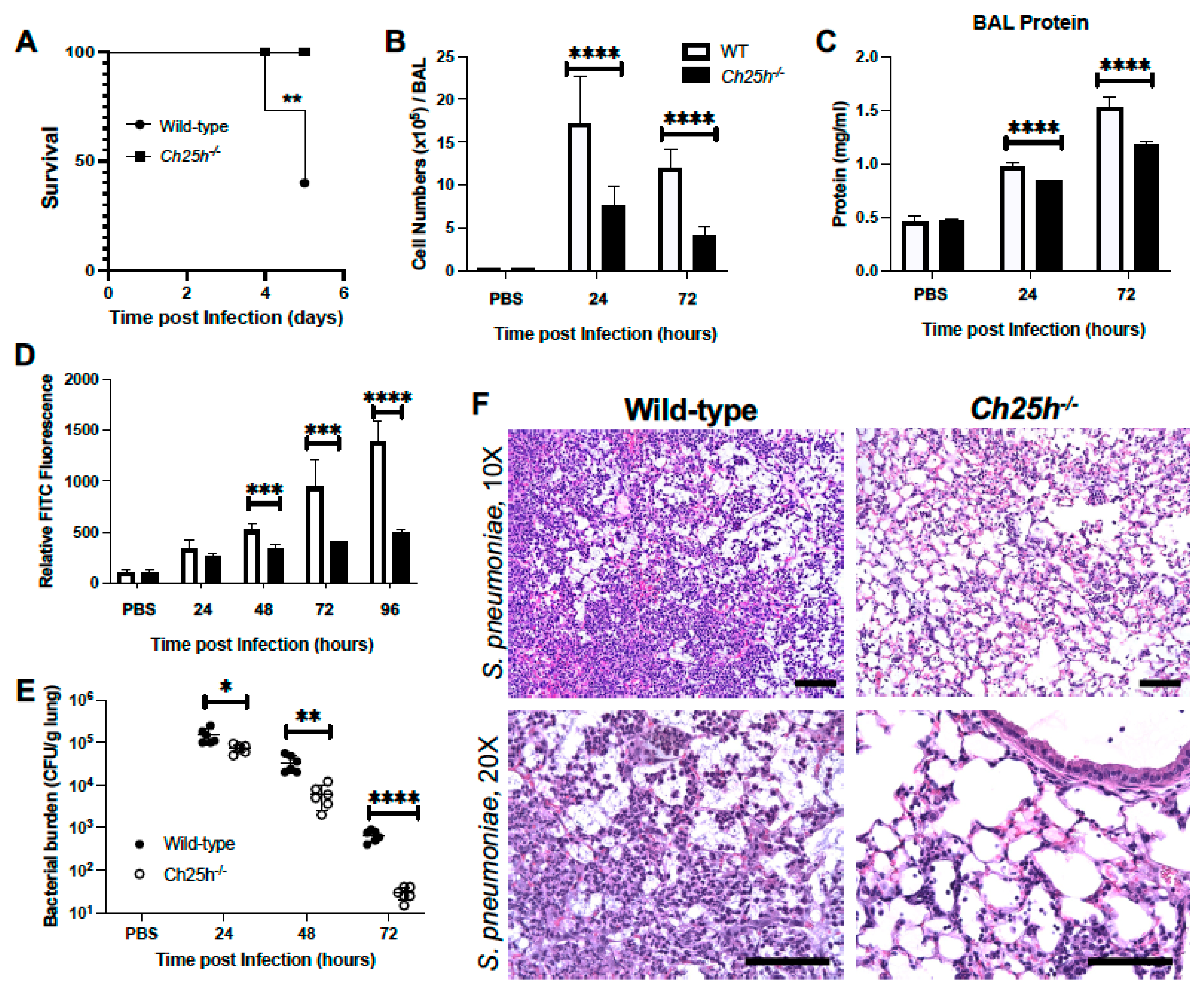
3.1. Decreased Lung Injury in Ch25h−/− Mice during S. pneumoniae Infection
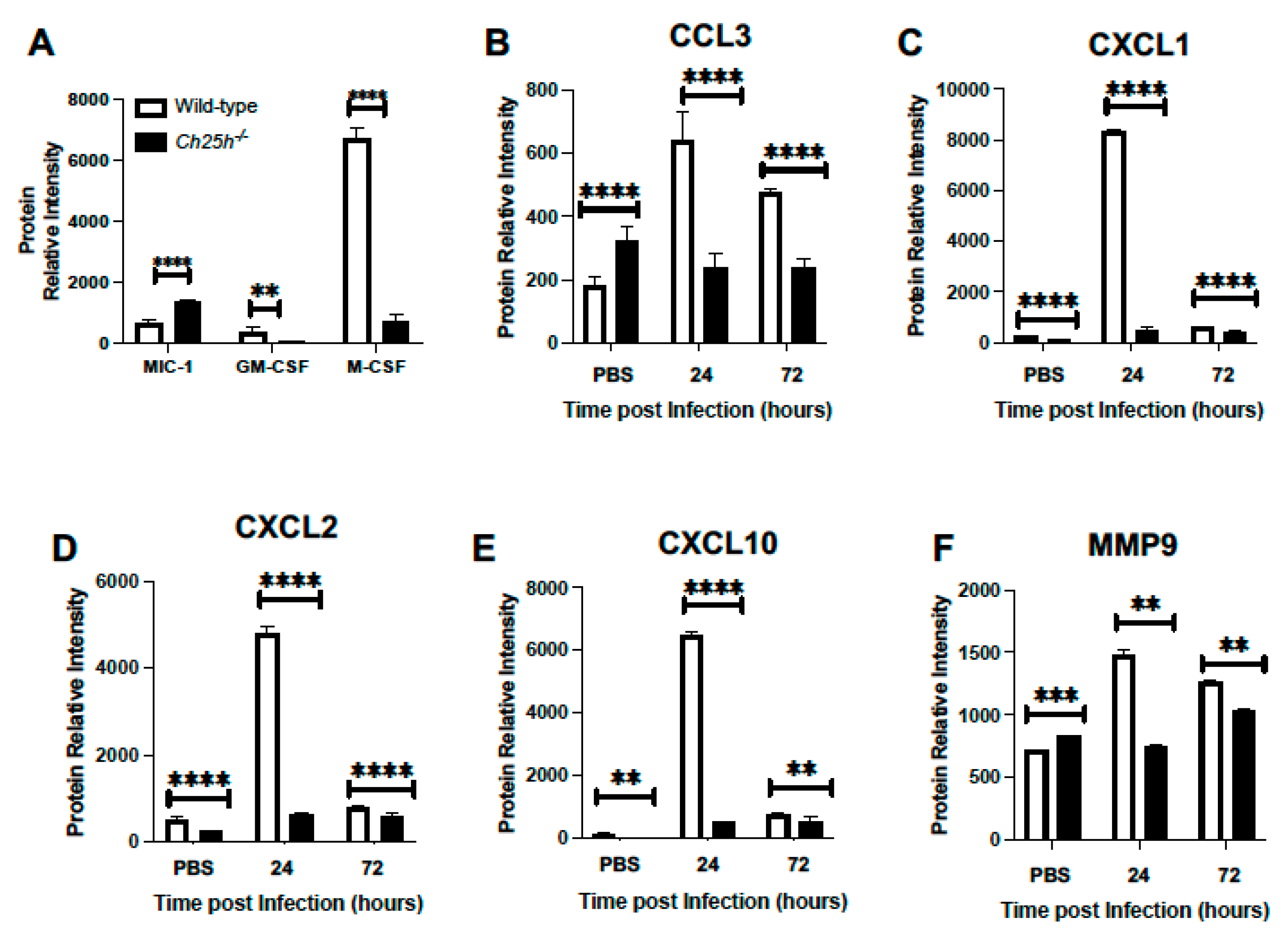
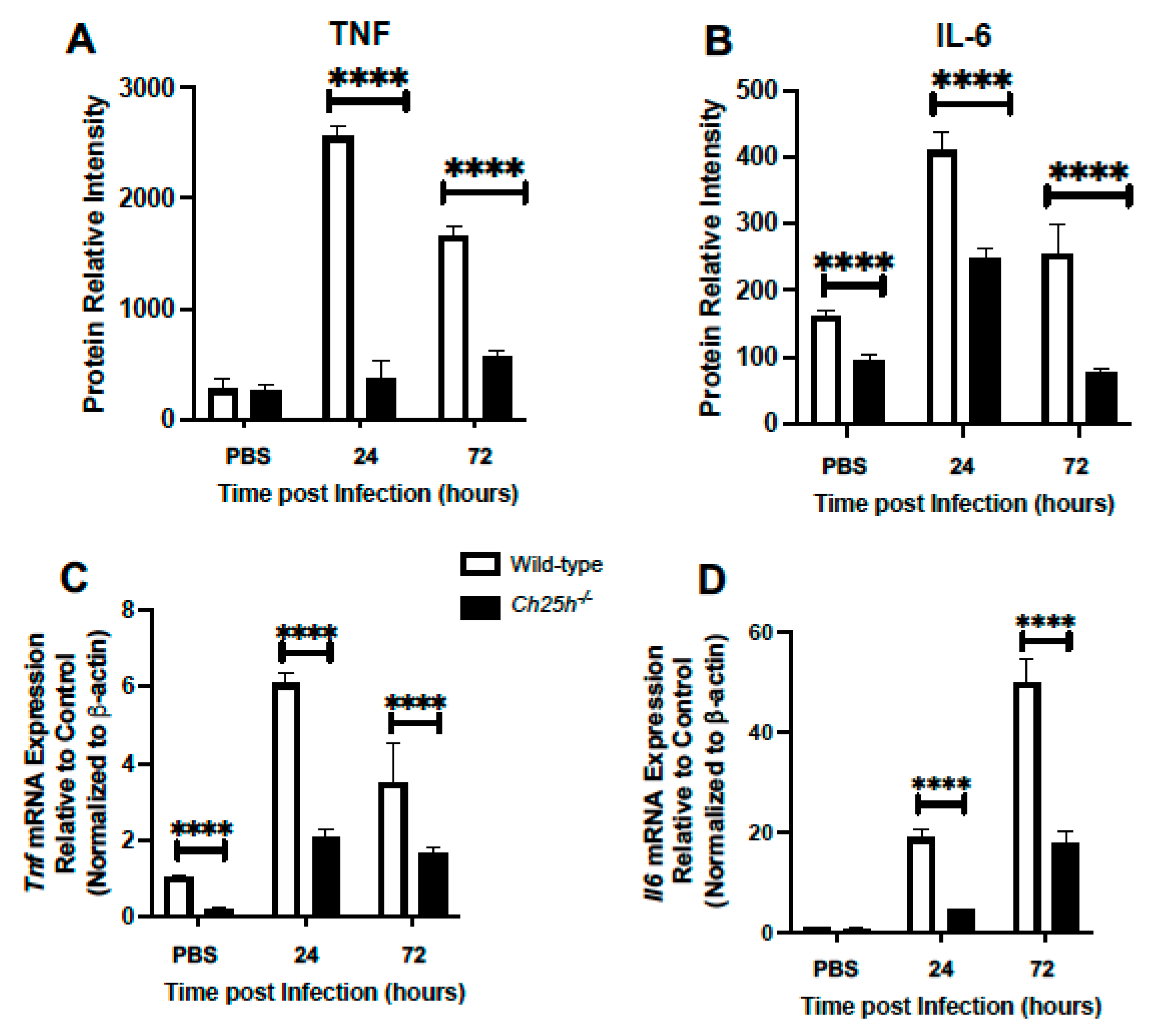
3.2. Decreased Chemokine and Inflammatory Cytokine Expression in Ch25h−/− Lung during S. pneumoniae Infection
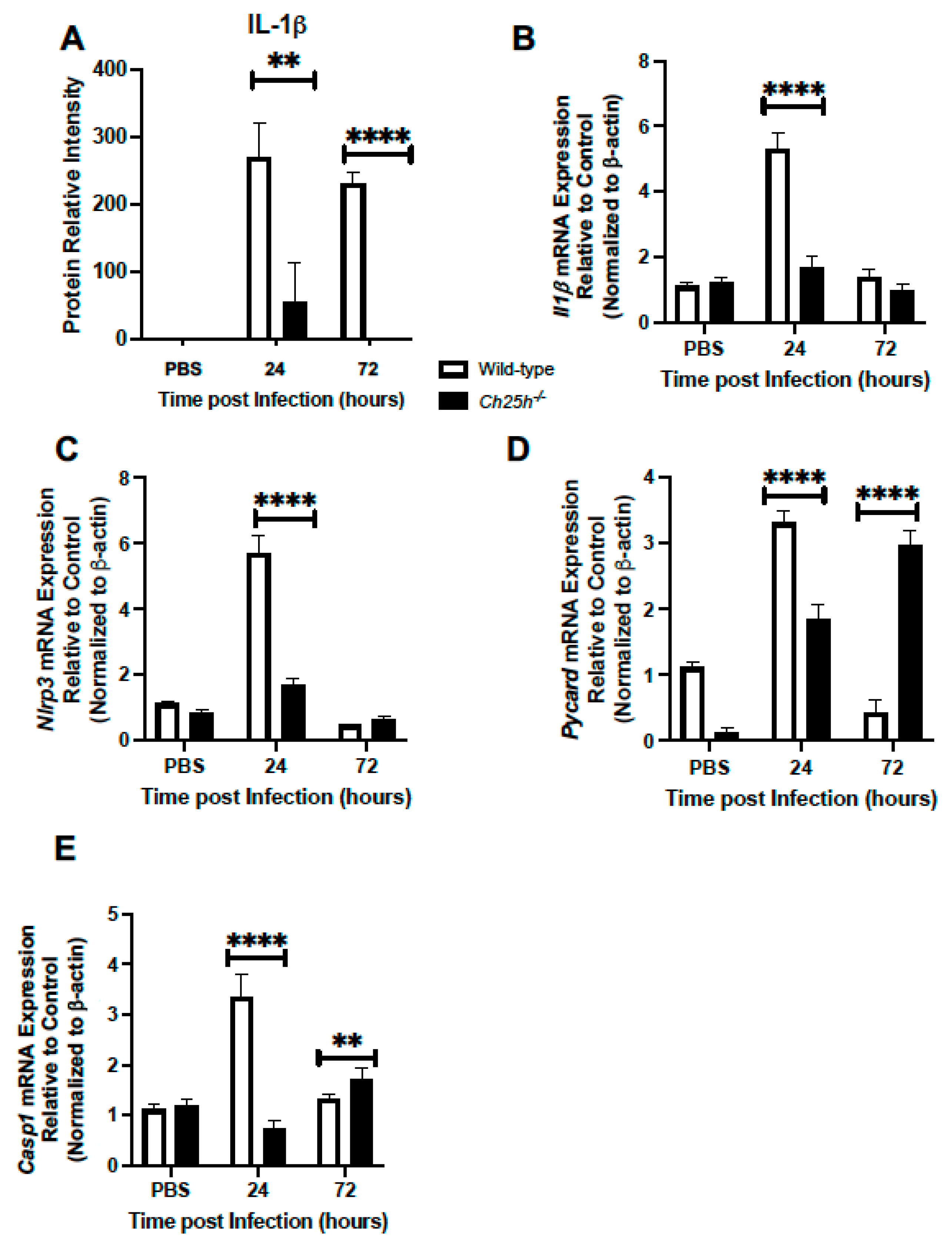
3.3. Decreased IL-1β Production and Inflammasome Gene Expression in Ch25h−/− Macrophages
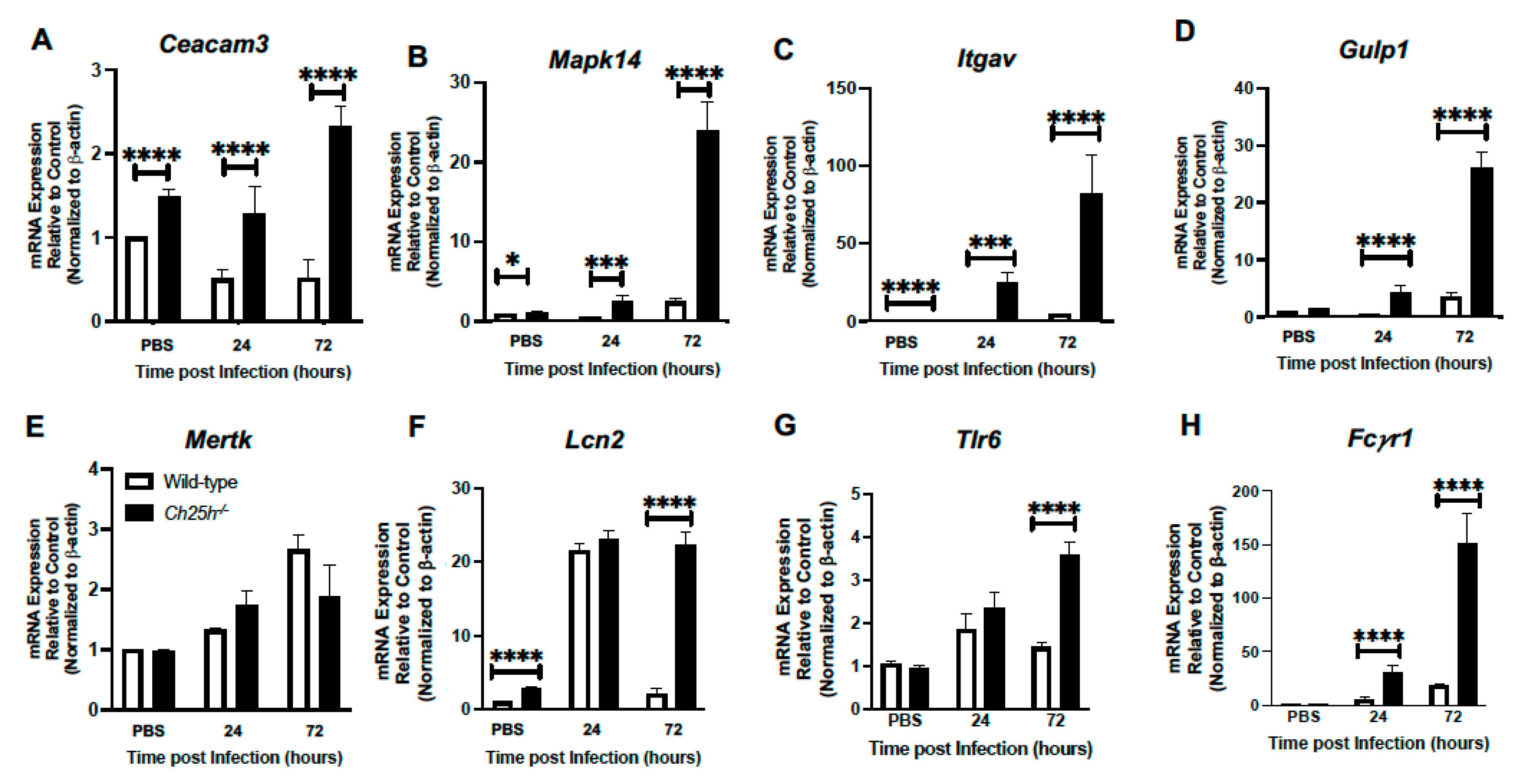
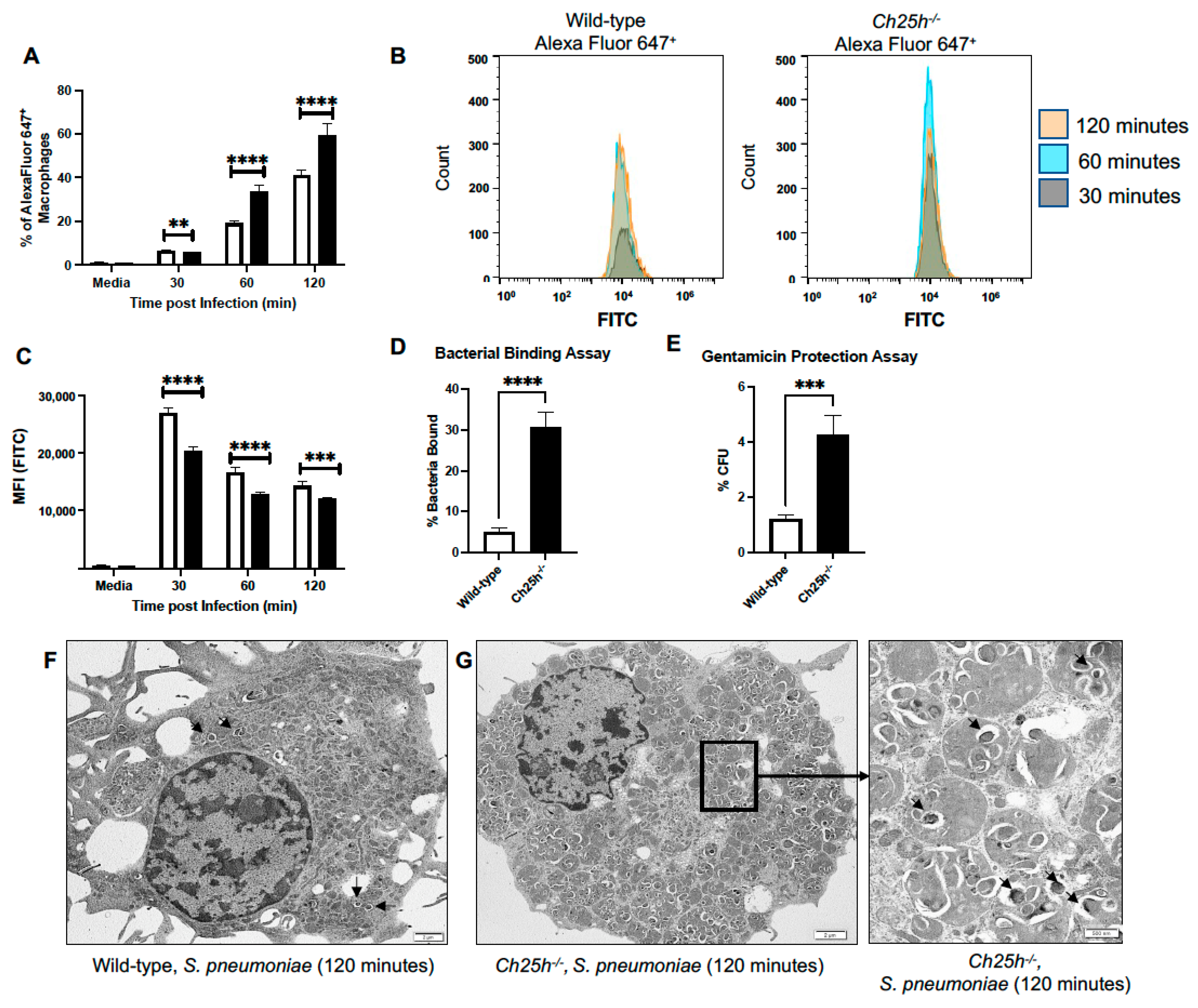
3.4. Enhanced Phagocytosis Receptor Expression by Ch25h−/− Macrophages during S. pneumoniae Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ortqvist, A.; Hedlund, J.; Kalin, M. Streptococcus pneumoniae: Epidemiology, risk factors, and clinical features. Semin. Respir. Crit. Care Med. 2005, 26, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Bernard, O.; Chun, L.; Croze, R.H.; Ross, J.T.; Nesseler, N.; Wu, X.; Abbott, J.; Fang, X.; Calfee, C.S.; et al. Clinically relevant model of pneumococcal pneumonia, ARDS, and nonpulmonary organ dysfunction in mice. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L717–L736. [Google Scholar] [CrossRef] [PubMed]
- Aberdein, J.D.; Cole, J.; Bewley, M.; Marriott, H.M.; Dockrell, D.H. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin. Exp. Immunol. 2013, 174, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Leemans, J.C.; Florquin, S.; Branger, J.; Maris, N.A.; Pater, J.; van Rooijen, N.; van der Poll, T. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 2003, 167, 171–179. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, V.R., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Afonso, M.S.; Machado, R.M.; Lavrador, M.S.; Quintao, E.C.R.; Moore, K.J.; Lottenberg, A.M. Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 2018, 10, 760. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Rawson, B.R.; Brown, S.M. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch. Biochem. Biophys. 2002, 397, 139–148. [Google Scholar] [CrossRef]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef] [Green Version]
- Diczfalusy, U.; Olofsson, K.E.; Carlsson, A.-M.; Gong, M.; Golenbock, D.T.; Rooyackers, O.; Fläring, U.; Björkbacka, H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J. Lipid Res. 2009, 50, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Karuna, R.; Christen, I.; Sailer, A.W.; Bitsch, F.; Zhang, J. Detection of dihydroxycholesterols in human plasma using HPLC-ESI-MS/MS. Steroids 2015, 99 Pt B, 131–138. [Google Scholar] [CrossRef]
- Madenspacher, J.H.; Morrell, E.D.; Gowdy, K.M.; McDonald, J.G.; Thompson, B.M.; Muse, G.W.; Martinez, J.; Thomas, S.Y.; Mikacenic, C.; Nick, J.A.; et al. Cholesterol 25-hydroxylase promotes efferocytosis and resolution of lung inflammation. JCI Insight 2020, 5, e137189. [Google Scholar] [CrossRef] [PubMed]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Kropp, K.A.; Forster, T.; Shui, G.; Lacaze, P.; Watterson, S.; Griffiths, S.J.; Spann, N.J.; et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 2013, 38, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Scott, L.A. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 2010, 88, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, W.; Wang, X.; Zhang, W.; Tian, H.; Deng, H.; Zhang, L.; Gao, G. Identification of new type I interferon-stimulated genes and investigation of their involvement in IFN-beta activation. Protein Cell 2018, 9, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hulten, L.M.; Wiklund, O. Macrophages isolated from human atherosclerotic plaques produce IL-8, and oxysterols may have a regulatory function for IL-8 production. Arter. Thromb. Vasc. Biol. 1997, 17, 317–323. [Google Scholar] [CrossRef]
- Lemaire-Ewing, S.; Berthier, A.; Royer, M.C.; Logette, E.; Corcos, L.; Bouchot, A.; Monier, S.; Prunet, C.; Raveneau, M.; Rébé, C.; et al. 7β-Hydroxycholesterol and 25-hydroxycholesterol-induced interleukin-8 secretion involves a calcium-dependent activation of c-fos via the ERK1/2 signaling pathway in THP-1 cells: Oxysterols-induced IL-8 secretion is calcium-dependent. Cell Biol. Toxicol. 2009, 25, 127–139. [Google Scholar] [CrossRef]
- Fu, H.; Spieler, F.; Großmann, J.; Riemann, D.; Larisch, M.; Hiebl, B.; Schlecht, K.; Jaschke, C.; Bartling, B.; Hofmann, B.; et al. Interleukin-1 potently contributes to 25-hydroxycholesterol-induced synergistic cytokine production in smooth muscle cell-monocyte interactions. Atherosclerosis 2014, 237, 443–452. [Google Scholar] [CrossRef]
- Pokharel, S.M.; Shil, N.K.; Gc, J.B.; Colburn, Z.T.; Tsai, S.-Y.; Segovia, J.A.; Chang, T.-H.; Bandyopadhyay, S.; Natesan, S.; Jones, J.C.R.; et al. Integrin activation by the lipid molecule 25-hydroxycholesterol induces a proinflammatory response. Nat. Commun. 2019, 10, 1482. [Google Scholar] [CrossRef] [Green Version]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Park, S.; Hur, H.J.; Cho, H.-J.; Hwang, I.; Kang, Y.P.; Im, I.; Lee, H.; Lee, E.; Yang, W.; et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun. 2016, 7, 13129. [Google Scholar] [CrossRef]
- Koarai, A.; Yanagisawa, S.; Sugiura, H.; Ichikawa, T.; Kikuchi, T.; Furukawa, K.; Akamatsu, K.; Hirano, T.; Nakanishi, M.; Matsunaga, K.; et al. 25-Hydroxycholesterol enhances cytokine release and Toll-like receptor 3 response in airway epithelial cells. Respir. Res. 2012, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Koarai, A.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Hirano, T.; Akamatsu, K.; Yanagisawa, S.; Furusawa, M.; Uno, Y.; et al. Increased 25-hydroxycholesterol concentrations in the lungs of patients with chronic obstructive pulmonary disease. Respirology 2012, 17, 533–540. [Google Scholar] [CrossRef]
- Liu, C.; Yang, X.V.; Wu, J.; Kuei, C.; Mani, N.S.; Zhang, L.; Yu, J.; Sutton, S.W.; Qin, N.; Banie, H.; et al. Oxysterols direct B-cell migration through EBI2. Nature 2011, 475, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Garifulin, O.; Berland, R.; Boyartchuk, V.L. Listeria monocytogenes infection induces prosurvival metabolic signaling in macrophages. Infect. Immun. 2011, 79, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Case, J.B.; Yutuc, E.; Ma, X.; Shen, S.; Castro, M.F.G.; Liu, Z.; Zeng, Q.; Zhao, H.; Son, J.; et al. 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc. Natl. Acad. Sci. USA 2020, 117, 32105–32113. [Google Scholar] [CrossRef]
- Wang, S.; Li, W.; Hui, H.; Tiwari, S.K.; Zhang, Q.; Croaler, B.A.; Rawlings, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020, 39, e106057. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, Z.; Tian, B.; Zhou, M.; Fu, Z.F.; Zhao, L. Cholesterol 25-hydroxylase suppresses rabies virus infection by inhibiting viral entry. Arch. Virol. 2019, 164, 2963–2974. [Google Scholar] [CrossRef]
- Zhou, Q.D.; Chi, X.; Lee, M.S.; Hsieh, W.Y.; Mkrtchyan, J.; Feng, A.-C.; He, C.; York, A.G.; Bui, V.L.; Kronenberger, E.B.; et al. Interferon-mediated reprogramming of membrane cholesterol to evade bacterial toxins. Nat. Immunol. 2020, 21, 746–755. [Google Scholar] [CrossRef]
- Bottemanne, P.; Paquot, A.; Ameraoui, H.; Guillemot-Legris, O.; Alhouayek, M.; Muccioli, G.G. 25-Hydroxycholesterol metabolism is altered by lung inflammation, and its local administration modulates lung inflammation in mice. FASEB J. 2021, 35, e21514. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.S.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Xiao, G.; Qu, Z. Murine Bronchoalveolar Lavage. Bio. Protoc. 2017, 7, e2287. [Google Scholar] [CrossRef] [PubMed]
- Halbower, A.C.; Mason, R.J.; Abman, S.H.; Tuder, R.M. Agarose infiltration improves morphology of cryostat sections of lung. Lab. Investig. 1994, 71, 149–153. [Google Scholar]
- Reboldi, A.; Dang, E.V.; Mcdonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef]
- Domon, H.; Oda, M.; Maekawa, T.; Nagai, K.; Takeda, W.; Terao, Y. Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Sci. Rep. 2016, 6, 38013. [Google Scholar] [CrossRef] [PubMed]
- Abrams, M.E.; Johnson, K.A.; Perelman, S.S.; Zhang, L.-S.; Endapally, S.; Mar, K.B.; Thompson, B.M.; McDonald, J.G.; Schoggins, J.W.; Radhakrishnan, A.; et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat. Microbiol. 2020, 5, 929–942. [Google Scholar] [CrossRef]
- van Lieshout, M.H.P.; de Vos, A.F.; Dessing, M.C.; de Porto, A.P.N.A.; de Boer, O.J.; de Beer, R.; Terpstra, S.; Florquin, S.; van’t Veer, C.; van der Poll, T. ASC and NLRP3 impair host defense during lethal pneumonia caused by serotype 3 Streptococcus pneumoniae in mice. Eur. J. Immunol. 2018, 48, 66–79. [Google Scholar] [CrossRef]
- Phipps, J.C.; Aronoff, D.M.; Curtis, J.L.; Goel, D.; O’Brien, E.; Mancuso, P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect. Immun. 2010, 78, 1214–1220. [Google Scholar] [CrossRef]
- Cruz, C.S.D.; Liu, W.; He, C.H.; Jacoby, A.; Gornitzky, A.; Ma, B.; Flavell, R.; Lee, C.G.; Elias, J.A. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe 2012, 12, 34–46. [Google Scholar]
- Paudel, S.; Baral, P.; Ghimire, L.; Bergeron, S.; Jin, L.; Decorte, J.A.; Le, J.T.; Cai, S.; Jeyaseelan, S. CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by Streptococcus pneumoniae serotype 3. Blood 2019, 133, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Spreer, A.; Kerstan, H.; Böttcher, T.; Gerber, J.; Siemer, A.; Zysk, G.; Mitchell, T.J.; Eiffert, H.; Nau, R. Reduced release of pneumolysin by Streptococcus pneumoniae in vitro and in vivo after treatment with nonbacteriolytic antibiotics in comparison to ceftriaxone. Antimicrob. Agents Chemother. 2003, 47, 2649–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetter, M.K. Serotypic variations among virulent pneumococci in deposition and degradation of covalently bound C3b: Implications for phagocytosis and antibody production. J. Infect. Dis. 1986, 153, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Neeleman, C.; Geelen, S.P.; Aerts, P.C.; Daha, M.R.; Mollnes, T.E.; Roord, J.J.; Posthuma, G.; Van Dijk, H.; Fleer, A. Resistance to both complement activation and phagocytosis in type 3 pneumococci is mediated by the binding of complement regulatory protein factor H. Infect. Immun. 1999, 67, 4517–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.J.; Pronko, A.; Yang, J.; Pagan, K.; Stout-Delgado, H. Role of Cholesterol 25-Hydroxylase (Ch25h) in Mediating Innate Immune Responses to Streptococcus pneumoniae Infection. Cells 2023, 12, 570. https://doi.org/10.3390/cells12040570
Cho SJ, Pronko A, Yang J, Pagan K, Stout-Delgado H. Role of Cholesterol 25-Hydroxylase (Ch25h) in Mediating Innate Immune Responses to Streptococcus pneumoniae Infection. Cells. 2023; 12(4):570. https://doi.org/10.3390/cells12040570
Chicago/Turabian StyleCho, Soo Jung, Alexander Pronko, Jianjun Yang, Kassandra Pagan, and Heather Stout-Delgado. 2023. "Role of Cholesterol 25-Hydroxylase (Ch25h) in Mediating Innate Immune Responses to Streptococcus pneumoniae Infection" Cells 12, no. 4: 570. https://doi.org/10.3390/cells12040570