
Prophylactic Glatiramer Acetate Treatment Positively Attenuates Spontaneous Opticospinal Encephalomyelitis

Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Flow Cytometry
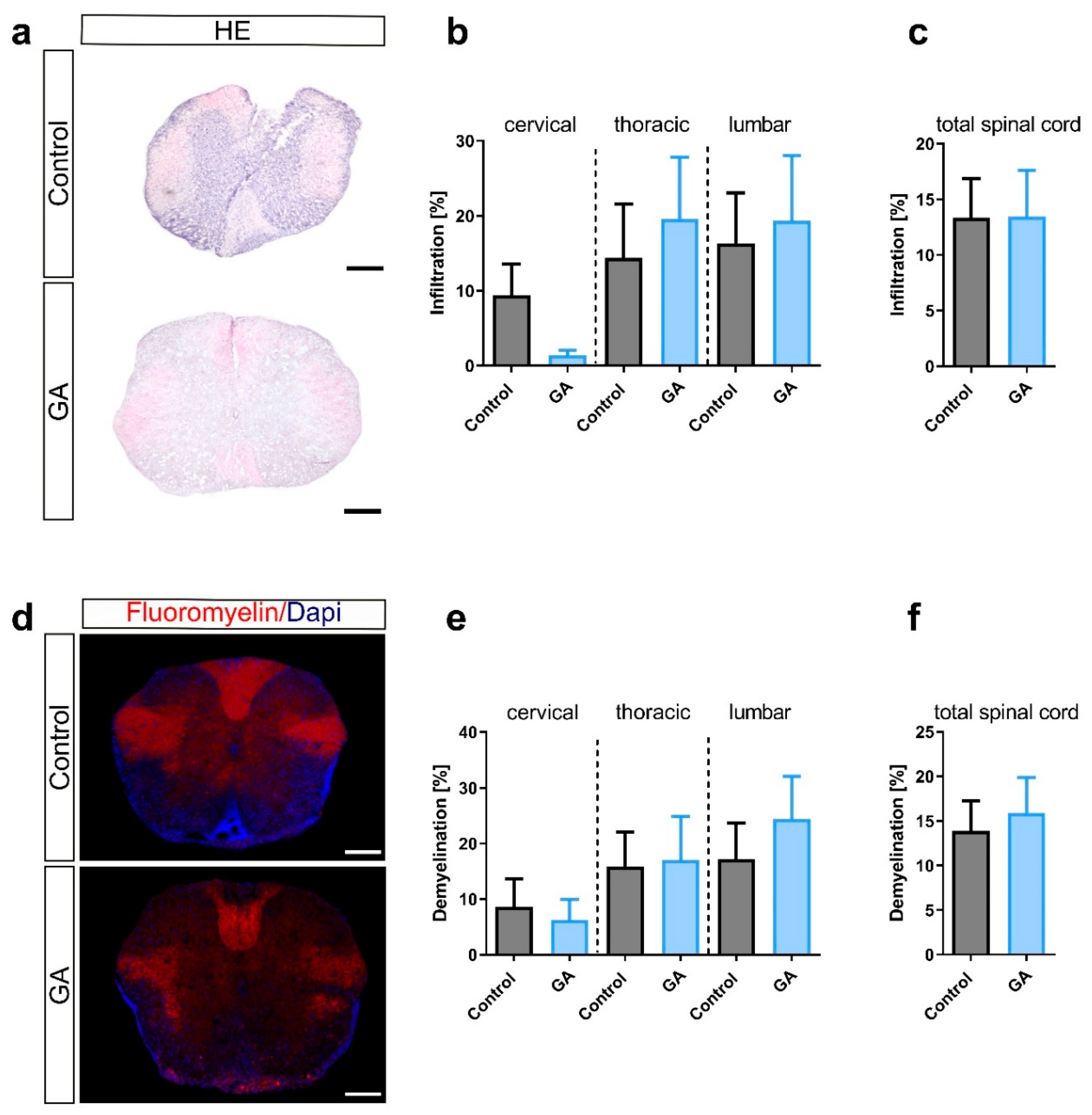
2.3. Histological Analyses
2.4. Immunocytochemistry and Microscopy
2.5. Statistical Analysis
3. Results
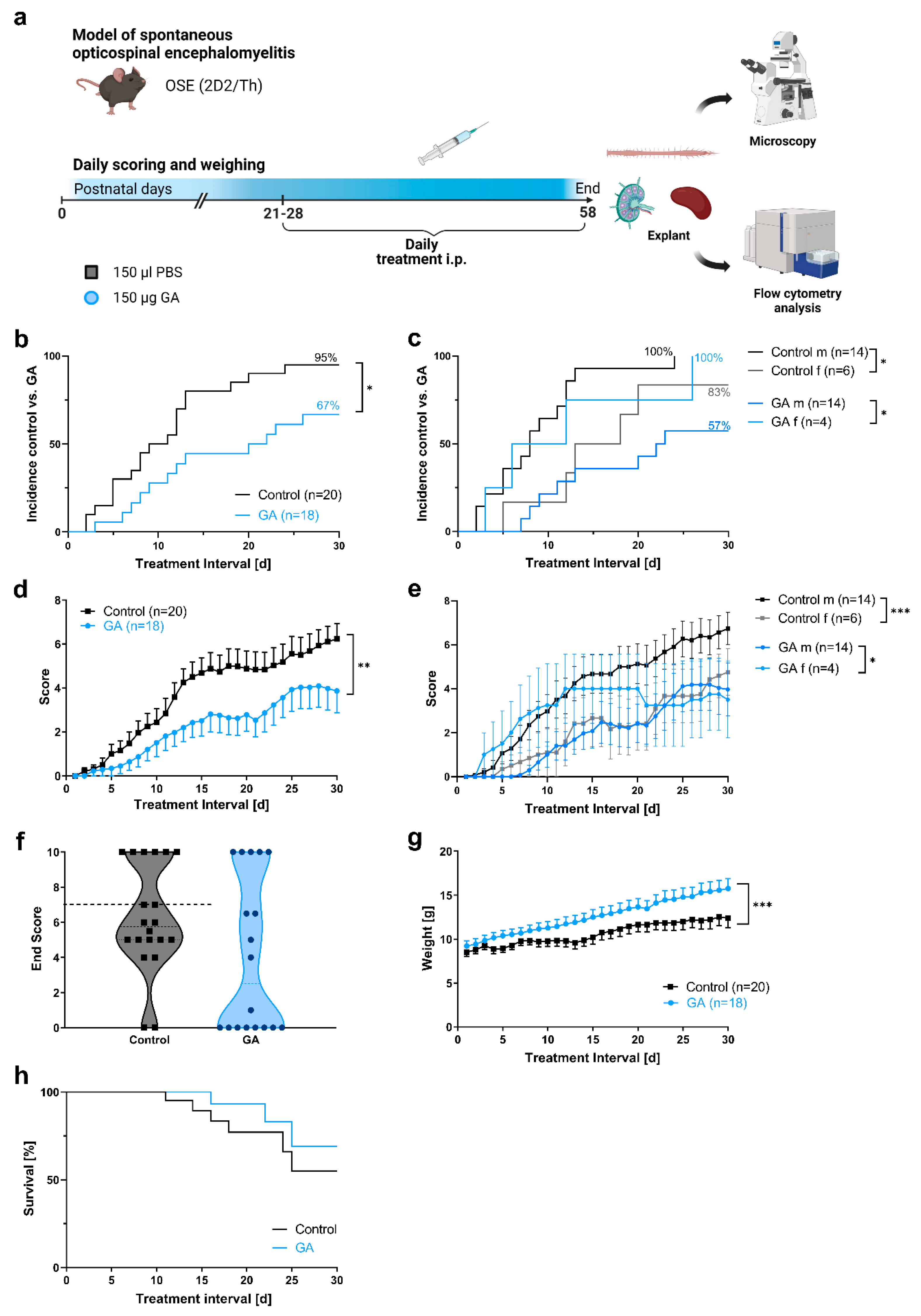
3.1. Prophylactic GA Treatment Improves Clinical Course and Body Weight in a Spontaneous Model of Autoimmune Encephalomyelitis
3.2. Prophylactic GA Treatment Slightly Reduces Overall Microglia/Macrophage Infiltration
3.3. Prophylactic GA Treatment Differentially Affects Lymphocyte Subpopulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple sclerosis-a quiet revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Borisow, N.; Mori, M.; Kuwabara, S.; Scheel, M.; Paul, F. Diagnosis and Treatment of NMO Spectrum Disorder and MOG-Encephalomyelitis. Front. Neurol. 2018, 9, 888. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Gold, R. Progressive multiple sclerosis: Latest therapeutic developments and future directions. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419878323. [Google Scholar] [CrossRef] [PubMed]
- Liblau, R. Glatiramer acetate for the treatment of multiple sclerosis: Evidence for a dual anti-inflammatory and neuroprotective role. J. Neurol. Sci. 2009, 287 (Suppl. S1), S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Boster, A.L.; Ford, C.C.; Neudorfer, O.; Gilgun-Sherki, Y. Glatiramer acetate: Long-term safety and efficacy in relapsing-remitting multiple sclerosis. Expert Rev. Neurother. 2015, 15, 575–586. [Google Scholar] [CrossRef]
- Prod’homme, T.; Zamvil, S.S. The Evolving Mechanisms of Action of Glatiramer Acetate. Cold Spring Harb. Perspect. Med. 2019, 9, a029249. [Google Scholar] [CrossRef]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef]
- Haupeltshofer, S.; Leichsenring, T.; Berg, S.; Pedreiturria, X.; Joachim, S.C.; Tischoff, I.; Otte, J.M.; Bopp, T.; Fantini, M.C.; Esser, C.; et al. Smad7 in intestinal CD4+ T cells determines autoimmunity in a spontaneous model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 25860–25869. [Google Scholar] [CrossRef]
- Petrikowski, L.; Reinehr, S.; Haupeltshofer, S.; Deppe, L.; Graz, F.; Kleiter, I.; Dick, H.B.; Gold, R.; Faissner, S.; Joachim, S.C. Progressive Retinal and Optic Nerve Damage in a Mouse Model of Spontaneous Opticospinal Encephalomyelitis. Front. Immunol. 2022, 12, 759389. [Google Scholar] [CrossRef]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, T.; Fassler, R.; Bauer, J.; Lassmann, H.; Linington, C.; Wekerle, H.; Iglesias, A. B lymphocytes producing demyelinating autoantibodies: Development and function in gene-targeted transgenic mice. J. Exp. Med. 1998, 188, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Ruck, T.; Gobel, K.; Henschel, C.; Afzali, A.M.; Gob, E.; Muntefering, T.; Kleinschnitz, C.; Wiendl, H.; Meuth, S.G. Effects of glatiramer acetate in a spontaneous model of autoimmune neuroinflammation. Am. J. Pathol. 2014, 184, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Hestvik, A.L.; Skorstad, G.; Price, D.A.; Vartdal, F.; Holmoy, T. Multiple sclerosis: Glatiramer acetate induces anti-inflammatory T cells in the cerebrospinal fluid. Mult. Scler. J. 2008, 14, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Korporal, M.; Balint, B.; Fritzsching, B.; Schwarz, A.; Wildemann, B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4+CD25+FOXP3+CD31+ T-cells in patients with multiple sclerosis. J. Neuroimmunol. 2009, 216, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Tyler, A.F.; Mendoza, J.P.; Firan, M.; Karandikar, N.J. CD8+ T Cells Are Required for Glatiramer Acetate Therapy in Autoimmune Demyelinating Disease. PLoS ONE 2013, 8, e66772. [Google Scholar] [CrossRef] [PubMed]
- Begum-Haque, S.; Sharma, A.; Christy, M.; Lentini, T.; Ochoa-Reparaz, J.; Fayed, I.F.; Mielcarz, D.; Haque, A.; Kasper, L.H. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2010, 219, 47–53. [Google Scholar] [CrossRef]
- Weber, M.S.; Starck, M.; Wagenpfeil, S.; Meinl, E.; Hohlfeld, R.; Farina, C. Multiple sclerosis: Glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004, 127, 1370–1378. [Google Scholar] [CrossRef]
- Weber, M.S.; Prod’homme, T.; Youssef, S.; Dunn, S.E.; Rundle, C.D.; Lee, L.; Patarroyo, J.C.; Stuve, O.; Sobel, R.A.; Steinman, L.; et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat. Med. 2007, 13, 935–943. [Google Scholar] [CrossRef]
- Molnarfi, N.; Prod’homme, T.; Schulze-Topphoff, U.; Spencer, C.M.; Weber, M.S.; Patarroyo, J.C.; Lalive, P.H.; Zamvil, S.S. Glatiramer acetate treatment negatively regulates type I interferon signaling. Neurol. Neuroimmunol. Neuroinflammation 2015, 2, e179. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Siglienti, I.; Grauer, O.; Magnus, T.; Scarlato, G.; Toyka, K. Induction of IL-10 in rat peritoneal macrophages and dendritic cells by glatiramer acetate. J. Neuroimmunol. 2004, 148, 63–73. [Google Scholar] [CrossRef]
- Chabot, S.; Yong, F.P.; Le, D.M.; Metz, L.M.; Myles, T.; Yong, V.W. Cytokine production in T lymphocyte-microglia interaction is attenuated by glatiramer acetate: A mechanism for therapeutic efficacy in multiple sclerosis. Mult. Scler. J. 2002, 8, 299–306. [Google Scholar] [CrossRef]
- Pul, R.; Moharregh-Khiabani, D.; Skuljec, J.; Skripuletz, T.; Garde, N.; Voss, E.V.; Stangel, M. Glatiramer acetate modulates TNF-alpha and IL-10 secretion in microglia and promotes their phagocytic activity. J. Neuroimmune Pharmacol. 2011, 6, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.L.; Coles, A.; Horakova, D.; Havrdova, E.; Izquierdo, G.; Prat, A.; Girard, M.; Duquette, P.; Trojano, M.; Lugaresi, A.; et al. Association of Initial Disease-Modifying Therapy with Later Conversion to Secondary Progressive Multiple Sclerosis. JAMA 2019, 321, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hummert, M.W.; et al. MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflammation 2016, 13, 280. [Google Scholar] [CrossRef] [PubMed]
- Ayzenberg, I.; Schollhammer, J.; Hoepner, R.; Hellwig, K.; Ringelstein, M.; Aktas, O.; Kumpfel, T.; Krumbholz, M.; Trebst, C.; Paul, F.; et al. Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: A multicenter retrospective study. J. Neurol. 2016, 263, 575–582. [Google Scholar] [CrossRef]
- Stellmann, J.P.; Krumbholz, M.; Friede, T.; Gahlen, A.; Borisow, N.; Fischer, K.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Immunotherapies in neuromyelitis optica spectrum disorder: Efficacy and predictors of response. J. Neurol. Neurosurg. Psychiatry 2017, 88, 639–647. [Google Scholar] [CrossRef]
- Gold, S.M.; Willing, A.; Leypoldt, F.; Paul, F.; Friese, M.A. Sex differences in autoimmune disorders of the central nervous system. Semin. Immunopathol. 2019, 41, 177–188. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Sex | Disease Incidence (%) | Incidence Depending on Sex | Mean Score ± SEM | Max Score ± SEM | Mean Day of Onset ± SEM * | Survival (%) | |
|---|---|---|---|---|---|---|---|
| GA | m 14 f 4 | 12/18 (67) | m 57% f 100% | 2.2 ± 0.57 | 4.4 ± 0.94 | 13.3 ± 2.19 | 13/18 (72.2) |
| Control | m 14 f 6 | 19/20 (95.0) | m 100% f 83% | 3.7 ± 0.51 | 6.5 ± 0.62 | 9.9 ± 1.39 | 14/20 (70.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koc, Ü.; Haupeltshofer, S.; Klöster, K.; Demir, S.; Gold, R.; Faissner, S. Prophylactic Glatiramer Acetate Treatment Positively Attenuates Spontaneous Opticospinal Encephalomyelitis. Cells 2023, 12, 542. https://doi.org/10.3390/cells12040542
Koc Ü, Haupeltshofer S, Klöster K, Demir S, Gold R, Faissner S. Prophylactic Glatiramer Acetate Treatment Positively Attenuates Spontaneous Opticospinal Encephalomyelitis. Cells. 2023; 12(4):542. https://doi.org/10.3390/cells12040542
Chicago/Turabian StyleKoc, Ümmügülsüm, Steffen Haupeltshofer, Katharina Klöster, Seray Demir, Ralf Gold, and Simon Faissner. 2023. "Prophylactic Glatiramer Acetate Treatment Positively Attenuates Spontaneous Opticospinal Encephalomyelitis" Cells 12, no. 4: 542. https://doi.org/10.3390/cells12040542