
The Role of TP53 in Adaptation and Evolution

1
Department of Basic and Clinical Sciences, University of Nicosia Medical School, 2414 Nicosia, Cyprus
2
School of Veterinary Medicine, University of Nicosia, 2414 Nicosia, Cyprus
*
Author to whom correspondence should be addressed.
Cells 2023, 12(3), 512; https://doi.org/10.3390/cells12030512
Submission received: 29 December 2022
/
Revised: 28 January 2023
/
Accepted: 31 January 2023
/
Published: 3 February 2023
(This article belongs to the Special Issue Role of TP53 Gene in Preventing Cancer and Promoting Adaptation)
{kind=link}
{kind=link}
Abstract
:The TP53 gene is a major player in cancer formation, and it is considered the most important tumor suppressor gene. The p53 protein acts as a transcription factor, and it is involved in DNA repair, senescence, cell-cycle control, autophagy, and apoptosis. Beyond cancer, there is evidence that TP53 is associated with fertility, aging, and longevity. Additionally, more evidence exists that genetic variants in TP53 are associated with environmental adaptation. Special TP53 amino-acid residues or pathogenic TP53 mutations seem to be adaptive for animals living in hypoxic and cold environments or having been exposed to starvation, respectively. At the somatic level, it has recently been proven that multiple cancer genes, including TP53, are under positive selection in healthy human tissues. It is not clear why these driver mutations do not transform these tissues into cancerous ones. Other studies have shown that elephants have multiple TP53 copies, probably this being the reason for the very low cancer incidence in these large animals. This may explain the famous Peto’s paradox. This review discusses in detail the multilevel role of TP53 in adaptation, according to the published evidence. This role is complicated, and it extends from cells to individuals and to populations.
1. Introduction
The TP53 gene encodes for the p53 protein, one of the main regulators of cell division and cell death. It is activated and stabilized after a variety of stresses. These stresses include radiation, toxic substances, hypoxia, high production of Reactive Oxygen Species (ROS), uncontrolled cell cycle, and others [1]. P53 can effectively trigger cell cycle arrest, DNA repair, senescence, apoptosis, and autophagy. Mutations in TP53 are found approximately in 50% of tumors. These mutations are under positive selection since they are highly beneficial for cell survival and proliferation [2,3,4].
There is an increasing amount of knowledge related to the evolution of TP53. Population selective pressures on TP53 are related to cancer, infection defense, and environmental stresses. Selective pressures at the somatic level may have a variety of effects. They can be harmful (cancer), protective, or of unknown significance. Molecular evolution research contributes to a deeper understanding of TP53 function and generally of the wider function of tumor suppressor genes. In this review, we summarize the current knowledge about TP53 evolution, focusing on the adaptational role of TP53.
2. Evolution of p53 Family
The gene family of TP53 includes TP53, TP63, and TP73. The three genes share a high sequence similarity, and they act as transcription factors. Each encoded protein has three DNA binding domains [3]. The high-level conservation of p53, p63, and p73 proteins in animals reveals the significance of their function. However, plants, fungi, eubacteria, and archaea do not contain protein sequences related to the p53 family (Figure 1). Choanoflagellates have a p53 orthologue; its DNA-binding domain and C terminus are very similar to the human p53 ones [5]. The common ancestor of the choanoflagellates and humans is probably one of the first multicellular organisms, estimated to have lived 600–800 million years ago. Interestingly, animals that have evolved at later times (anemones, flies, and nematodes) all have functional proteins with high sequence similarity to the human p53 DNA-binding domains [3].
3. Role of TP53 Variants in Increased Fertility and Longevity
3.1. Fertility and TP53
P53 family members, p53, p63, and p73, have an important role in fertility and embryonic development [6]. Here, we are going to summarize the role of p53 on fertility and reproduction, under the view of evolution and adaptation.
TP53-R72P is a widely studied genetic variant. It is not too clear how the 72 alleles’ frequencies are regulated by natural selection [7]. The R72 variant can induce apoptosis more effectively than the P72 one [8]. The proline allele (P) is most frequent in individuals of African descent, and the arginine allele (R) is most frequent in Caucasians. The 72 residue belongs in a proline-rich domain of p53, where a significant number of mutations has been reported [9]. A combination of alleles in this region may predispose to cancer [9].
Kang et al. (2009) [10] showed that the P72 variant is significantly enriched in an in vitro fertilization (IVF) patients’ group, especially in patients who are younger than 35 years of age. They showed that young patients, being homozygous for the P72 allele, have higher implantation and pregnancy failure rates after IVF, compared to the patients carrying at least one R72 allele. They also found evidence that the P72 effect is performed by lowering the leukemia inhibitor factor (LIF), an essential protein for embryo implantation. P53 regulates transcription of LIF through a DNA response element that is found in intron 1 of LIF gene [11]. Lledo et al. (2014) [12] found similar results. They showed that P72 is a risk factor for recurrent implantation failure and recurrent pregnancy loss. Women having the P72 allele in homozygosity have a lower rate of embryo implantation and eventually a lower pregnancy rate, in comparison with the R72 carriers [12]. Lledo et al. (2014) [12] concluded that P72 is involved in human fertility and that genotyping of the TP53 gene could potentially determine the prognosis of IVF cycles. A meta-analysis and systematic-review study, incorporating many studies, found that recurrent pregnancy loss is associated with homozygous P72 women, most notably in Caucasians [13]. The association of the P72 allele with pregnancy loss may be population specific.
Evidence was found that the TP53-P72 allele may have a role after implantation, enhancing the chance for a double pregnancy. Twins’ mothers in Candido Godoi, a town in Brazil, have been found to have an increased number of P72 alleles, making the P72 a main risk factor for twinning [14]. The authors speculate that this may be related to the reduced apoptosis potential of the P72 allele. If the association of twinning with the P72 alleles is true, then the molecular mechanism needs to be deciphered.
Interestingly, there is also some evidence that relates p53 null mice with a reproduction advantage. Heterozygous p53+/− male mice seem to achieve higher fertilization rates than controls. On the other hand, p53+/− female mice release higher number of oocytes than controls [15]. If these observations are correct, and they are also valid in the wild animal populations, this provides a tendency for accumulation of deleterious mutations within a population, increasing carcinogenesis risk. This can be viewed under the lens of the antagonistic pleiotropy phenomenon, where mutations that are harmful at old age were selected because they are beneficial (increase the Darwinian fitness) in earlier life [16].
3.2. Longevity and TP53
The potential contribution of TP53 to longevity has been studied in some animal models that will be analyzed in the next paragraphs. Evidence exists that p53 null alleles or p53 functional variants are related to longevity. The paradox is that these alleles also increase cancer risk or restrict normal p53 functions.
In Drosophila melanogaster, dominant negative versions of p53 in neurons extended adult life span and increased the resistance to oxidative stress in the fly [17]. Flies with a selective reduction of p53 activity in adult neurons were more resistant to paraquat poisoning than control flies. Interestingly, these flies had a normal female fecundity and a normal physical activity. These observations suggest that decreased p53 activity has positive effects on some aging phenotypes [17]. Activity regulation of Drosophila p53 seems to be like that of mammals. This regulation is performed by ubiquitination, phosphorylation, and sumoylation by multiple enzymes [18]. Another piece of evidence in Drosophila melanogaster is that the over-expression of p53 limits the life span in females, but favors the life span in males. On the other hand, another group showed that a p53 null mutation increased the life span of female flies, but it had less significant and more variable effects in males [19].
RNA interference or genetic knockout of the Caenorhabditis elegans p53 ortholog, cep-1, resulted in increased life span, depending on the daf-16 gene functionality [20]. Experiments by another group showed that a cep-1 deletion in Caenorhabditis elegans increased by 25% the lifespan of the worm [21]. In these experiments, the cep-1 gene was lacking the DNA-binding domain.
A study by van Heemst et al. (2005) [22] resulted in a paradox. They found that the TP53 72P/P genotype is associated with an increased cancer risk in humans when compared to the 72R/R genotype. However, when they analyzed 1226 people aged 85 years and over, they found that the 72P/P carriers had a 41% increased survival (p = 0.032), despite demonstrating a 2.54-fold (p = 0.007) proportional mortality risk from cancer [22]. Another study in the Danish population found an increased longevity for the 72R/P heterozygotes and 72P/P homozygous, when compared to the 72R/R homozygotes. This can be explained partly by the better prognosis after the diagnosis of cancer or other diseases. The cumulative 5-year mortality after cancer diagnosis was reduced, with an increasing number of P72 alleles. The analysis also showed that the P72 allele prolongs life on an average of 3 years [23]. Zhao et al. (2018) [24] investigated the effect of the p53 P72 allele in a mouse model. They proved that the P72 allele reduces p53 activity, increasing in this way the risk for tumor development in mice. However, mice that carry the P72 allele and escape tumor development display a longer lifespan than R72 mice and have a delayed development of aging-associated phenotypes. The authors also showed that the P72 allele influences aging and longevity through the better ability of p53 to retain and regulate the self-renewal function of stem cells, compared to the p53 R72 variant [24].
An interesting question is if Li-Fraumeni patients who carry heterozygous germ-line mutations in TP53 and survive cancer exhibit any increased lifespan traits. No such associations have been published so far. Additionally, genetic variants of the MDM2 gene, the main negative regulator of TP53, could potentially be associated with longevity. There is only one published study so far showing an association of an MDM2 SNP with longevity, but only for female individuals [25].
4. TP53 Is Crucial for Environmental Adaptation
P53 protein plays a fundamental physiological role in maintaining homeostasis by functioning as a transcription factor. P53 rapidly responds to many environmental stimuli by tetramer formation and then activation directly or indirectly a variety of genes. P53 monomers is the inactive state, and tetramers is the active state of p53 protein [26]. Its adaptive functions have an important impact on cell homeostasis, especially under changing environmental conditions caused by infections, metabolic alteration, or damage [27]. As was previously mentioned, p53 becomes activated in response to various stresses, including DNA damage, oncogene activation, oxidative stress, and hypoxia. This activation probably facilitates cellular cooperation in a coordinated manner, enabling the survival of the individual in a variety of environmental conditions. However, the validity of this assumption needs investigation.
The mole rat, Spalax ehrenbergi, since its origin 40 million years ago, has evolved adaptations to underground life in a highly hypoxic environment. A study revealed a special amino-acid residue in Spalax ehrenbergi p53 protein that is different form the conserved one in humans and mice. The amino-acid residue involves a substitution from arginine to lysine at the 174 position, in the p53 DNA-binding domain [28]. This residue is probably related to the reduced apoptosis potential of p53. This is in contrast with the fact that cancer occurrence is very rare in Spalax species. However, the same team showed that this Spalax p53 homologue can effectively induce autophagy, another type of cell death [29]. This altered function of p53 in Spalax is probably related to evolutionary adaptive mechanisms to hypoxia in underground life. It is very interesting that similar p53 amino-acid residues have been found in human tumors [28], this probably showing the adaptation of solid tumors in a hypoxic environment. Molecular similarities of Spalax response to hypoxia with tumor response to hypoxia are supported by published studies [30,31,32].
Zhao et al. (2013) [33] found similar results to those of Ashur-Fabian et al. (2004) [28] in some mammalian species living in a very high altitude in Tibet. Wild zokor Myospalax baileyi and root vole Microtus oeconomus have the p53 residues asparagine-104 (N104) and glutamic acid-104 (E104), respectively, differing from the serine-104 (S104) seen in other rodents, including the lowland subterranean zokor Myospalax cansus, and from the serine-106 (S106) in humans. More specifically, cells of Myospalax baileyi and Microtus oeconomus seem to be resistant to apoptosis under hypoxia, hypercapnia, and cold temperature [33]. These two different p53 codons are obviously an outcome of the environmental adaptation under specific ecological stresses found at the Tibet plateau. In human cancers, a germ-line mutation at this codon was reported in a patient with multiple primary cancers [34].
The subspecies Spalax galili, which lives in Israel, has underwent adaptive sympatric speciation due to divergent chalk and basalt ecologies of its environmental niche [35]. Higher methylation levels were noted on several sites of the p53 promoter in the population living in the chalk soil environment. In the same study, the authors showed that the diverse expression levels of p53 can affect cell-cycle arrest, but not the apoptotic mechanisms [35]. It is believed that methylation modification of p53 is an evolutionary adaptation under the environmental stresses of the abutting divergent chalk–basalt ecologies [35].
Axolotl (Ambystoma mexicanum, also known as Mexican salamander) is an amphibian that exhibits remarkable resistance to cancer development. Villiard et al. (2007) [36] compared axolotl p53 to the human one, and they found multiple different amino-acid residues. Interestingly, the axolotl special amino-acids have been found in human tumors [36]. Experiments showed that the activation and stability of axolotl p53 is temperature sensitive [36]. It seems that axolotl p53 variants have been selected as an adaptation to external oxygen or temperature variations.
The Andeans have adapted to the Altiplano in different ways than other natives living at high altitudes. Jacovas et al. (2018) [37] found evidence for positive selection on three genes, CLC, SP100, and DUOX2, in Native Americans living at a very high altitude in the Andean Artiplano. These genes are involved in the p53 pathway, and they are related to routes for the high-altitude hypoxia response. CLC encodes for a lysophospholipase that is expressed in eosinophils and basophils. DUOX2 is involved in a p53-dependent checkpoint for cell cycle entry. SP100 is a modulator of the p53 activity. The authors speculate that the genetic variants that are under selection on SP100 and DUOX2 genes contribute to a most efficient response of p53 under a hypoxic environment in high altitudes [37]. Under the same logic, the same group found that genetic variants on the MDM2, USP7, and LIF genes, encoding for proteins that are linked to the p53 transcriptional function, are probably under selection and play a significant role in the adaptation to the high altitudes of the Andean mountains [38]. Interestingly, Shi et al. (2009) [39] found that the p53 R72 allele is enriched in some East Asian human populations against the P72 allele, as an adaptation in cold winter temperatures. The authors argue that the selection of the R72 allele in cold environments is probably explained by the ability of p53 to activate mitochondrial respiration and provide more energy production during winter. Regulation of mitochondrial respiration by p53 is supported by previous studies [40]. They also found that a variant of the MDM2 gene (mdm2 is the primary negative regulator of the p53 protein) is associated with an adaptation under high UV radiation in high altitudes [39]. We remind here that the R72 variant makes p53 more effective to induce apoptosis in comparison to the P72 variant [8].
The adaptation potential of a missense carcinogenic mutation on TP53 gene was investigated in zebrafish, in fish larvae that were exposed under extreme starvation conditions [41]. The derived results showed that more mutated zebrafish larvae survived under extreme starvation conditions (no food at all) when compared to the wild-type ones. The experiment was performed under constant laboratory conditions, counting for larva fatalities until the 15th day post fertilization [41].
Another aspect of p53 evolution is viral infections, especially papillomavirus infections. Papillomaviruses can infect primates. Like many other viruses, papillomaviruses co-evolve with their hosts. E6 proteins of papillomaviruses stimulate polyubiquitination of p53 and subsequent proteosome-dependent degradation, thus predisposing the host for cancer [42]. Special amino-acid residues have been recognized on the host p53 and on the viral E6, being responsible for the host-pathogen-specific E6-mediated p53 degradation [43]. A study supported that non-human papillomaviruses have an adaptation toward a host carcinogenic phenotype [43]. Some macaque papillomavirus types exhibit a potential for cross-host p53 degradation, implying that complex pathogen–host interactions may exist [43].
5. Selection Pressures on Somatic TP53 Mutations
5.1. TP53 Mutations in Normal Tissues
It is well known that cancer is formed by the expansion of specific cell populations carrying the same DNA mutations, called mutational clones [44]. Driver mutations are the ones that are primarily responsible for the transformation of normal tissues into cancerous ones. Driver mutations are under positive selection since they offer an evolutionary advantage to cells. Studies have supported the hypothesis that somatic driver mutations being under selection exists also in normal tissues, sometimes in higher frequencies than in the malignant tissues [45]. This phenomenon is not fully understood, and it shows the complexity of tumor evolution from first mutation to a benign growth and, eventually, to cancer [44]. This research was highly expedited by the recent progress of genetics technologies, especially Next Generation Sequencing. In this section, we will focus on the available evidence about TP53 driver mutations in normal tissues.
Martincorena et al. (2015) [46] found that a surprisingly significant amount of cancer genes undergo strong positive selection in 18–32% of normal skin cells. The authors showed that aged sun-exposed skin has multiple evolving clones, and many of them carry a predisposition to cutaneous squamous cell carcinomas [46]. Interestingly, some healthy cells had 2–3 driver mutations. The same study showed that 3–5% of normal skin cells had TP53 mutations, and strong evidence was provided for clonal expansion of p53-mutated keratinocytes [46].
Martincorena et al. (2018) [47] studied the normal esophageal epithelium in different age donors. It was found that a strong positive selection is acted on clones carrying mutations in genes that predispose to cancer, and these mutations accumulate with age. Surprisingly, a significantly higher amount of NOTCH1 gene mutations was noted in normal esophageal epithelium, compared to the cancerous esophageal epithelium. TP53 driver mutations under selection were found in 5–10% of the normal esophageal epithelium of the heathy donors [47]. Colom et al. (2020) [48] supported the above findings by finding numerous mutant clones with multiple genes under positive selection in normal esophageal epithelium, including NOTCH1, NOTCH2, and TP53. Yokohama et al. (2019) [49] also found similar results in normal esophageal epithelium, including TP53 driver mutations under positive selection. The authors underline the fact that cancer mutations can be substantially accelerated by smoking and alcohol consumption. This excessive clonal expansion is implicated in cancer development, highlighting the importance of lifestyle factors in carcinogenesis [49].
The clonal expansions and somatic genetic changes of colorectal adenocarcinoma were also studied [50]. Some mutational processes were ubiquitous and continuous. Most mutations in colon crypts came from a single ancestral stem cell. Probable driver mutations under positive selection were present in around 1% of normal colorectal crypts. TP53 mutations were rare among these driver mutations in normal cells, in comparison to the colorectal cancer TP53 mutations that are found in ~56% of cases [50].
Carcinogens in cigarette smoke directly damage and mutate DNA, causing lung cancer. Yoshida et al. (2020) [51] studied the human bronchial epithelium and discovered that driver mutations increased in frequency with age, affecting 4–14% of cells in middle-aged never-smokers. In current smokers, ≥25% of cells carried driver mutations, and 0–6% cells had 2 or even 3 drivers. They used an algorithm to assess if any mutations are under positive selection in normal bronchial epithelium. Three genes were revealed through this algorithm: NOTCH1, TP53, and ARID2. It was clear through this study that tobacco smoking increases mutational burden and driver mutations. However, quitting smoking slows the accumulation of further damage in bronchial epithelium [51].
A study found somatic clonal expansion in morphologically normal urothelium [52]. More specifically, the chromatin remodeling genes KMT2 and KDM6A were the most commonly mutated in urothelial cells. TP53 driver mutations were found in 3.8% of the normal urothelial cells [52].
In conclusion, multiple studies show that healthy epithelial tissues accumulate cancer driver mutations with positive selection, a phenomenon that is accelerated with aging. A recent study gives some evidence why this happens. Colom et al. (2021) [53] showed that mutant clones in normal epithelium have an anti-tumorigenic role by competing and eradicating early tumors, preserving tissue integrity. Additionally, we could speculate that these mutations make tissues more resistant to harmful or challenging conditions. The continuous progress in single-cell sequencing technologies will definitely help for the better understanding of the role of somatic mutations in health, disease, aging, and evolution.
5.2. TP53 Mutations in Malignant Tissues
As previously mentioned, TP53 driver mutations is a common finding in human tumors. About 50% of all tumors have mutations in the TP53 gene [4]. However, TP53 mutations are not always the first mutations that appear in cancer cells. It is well known for some cancer types that TP53 mutations are selected when other cancer mutations already exist. It is not clear why this happens [3].
TP53 mutations are commonly found together with inherited and spontaneous BRCA1 or BRCA2 mutations in ovarian and breast tumors [54]. Remarkably, the 70–90% of cancers with BRCA1 mutations also have mutations in the TP53 gene [54]. This seems to be a more general phenomenon, since TP53 mutations frequently co-occur with mutations in DNA damage repair genes. However, the reason is still unknown.
TP53 mutations are frequently observed in many lung, colon, and pancreatic tumors with RAS mutations [54,55]. Earlier studies have shown that TP53 mutations can help KRAS-mutated cells to adaptively overcome replicative senescence. This may be the reason why TP53 mutations are found together with KRAS mutations [56].
6. TP53 Copy Numbers
Peto’s paradox is the observation that cancer incidence is not linearly associated with the number of cells of species [57,58,59]. This is a paradox since it is expected that a greater number of cells and cell divisions increases the chance of mutation accumulation and, consequently, the chance for malignancies. In 2015, Abegglen and colleagues [60] confirmed that elephants are cancer-resistant, despite their large body size and their long life span.
Abegglen et al. (2015) [60] found that elephants have at least 20 copies of TP53, although humans have only 1 copy. They also proved that elephants’ lymphocytes are more sensitive to apoptosis signaling in response to DNA damage from ionizing radiation when compared to the human ones. Elephants have a lower cancer rate and cancer mortality compared to humans. The multiple copies of TP53 and the enhanced p53-mediated apoptosis could represent an evolutionary strategy for cancer suppression [60]. Consistent with this observation, transgenic mice with multiple TP53 copies are significantly resistant to carcinogenesis [61].
Sulak et al. (2016) [62] expanded this research by including more elephant species in their study. They showed that most of the TP53 retrogene copies of elephants are transcribed and likely translated. These TP53 copies do not function as transcription factors, but they contribute to the enhanced sensitivity of elephant cells to DNA damage and the induction of apoptosis [62].
Padariya et al. (2022) [63] explained that elephant p53 isoforms have modified BOX-1 motifs to exhibit reduced binding capacity to mdm2 protein. P53 is regulated by the MDM2 E3 ubiquitin ligase. MDM2 is an oncoprotein and the main p53 regulator by blocking its transcriptional activity. Some species of elephants have been found to have TP53 copies that express a variety of BOX-1 MDM2-binding motifs, enhancing the sensitivity to cellular stresses [63]. This accounts for adaptations that favor healthy aging, such as cancer defenses. Mutations altering the structures of the MDM2-p53 partners have significant functional consequences for elephants, since cancer defense can be increased when p53 isoforms escape MDM2-mediated repression [63].
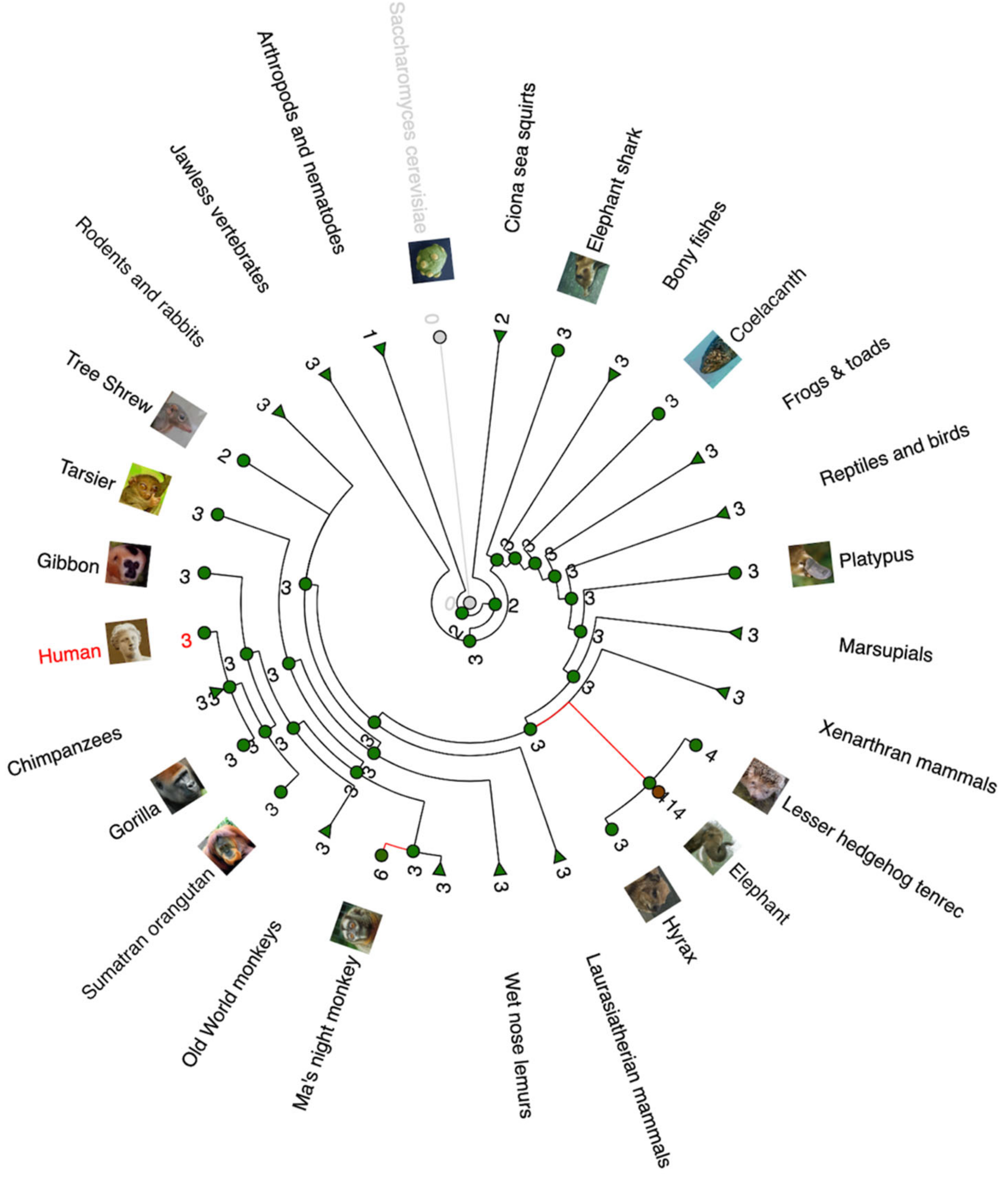
Analysis of TP53 copy numbers in multiple species can be seen in Figure 1 through a gain/loss phylogenetic tree illustration. It is obvious that most animal species have three TP53 paralogues in their genome, most likely TP53, TP63, and TP73. A remarkable copy expansion is observed in elephants and in Ma’s night monkey.
7. Conclusions
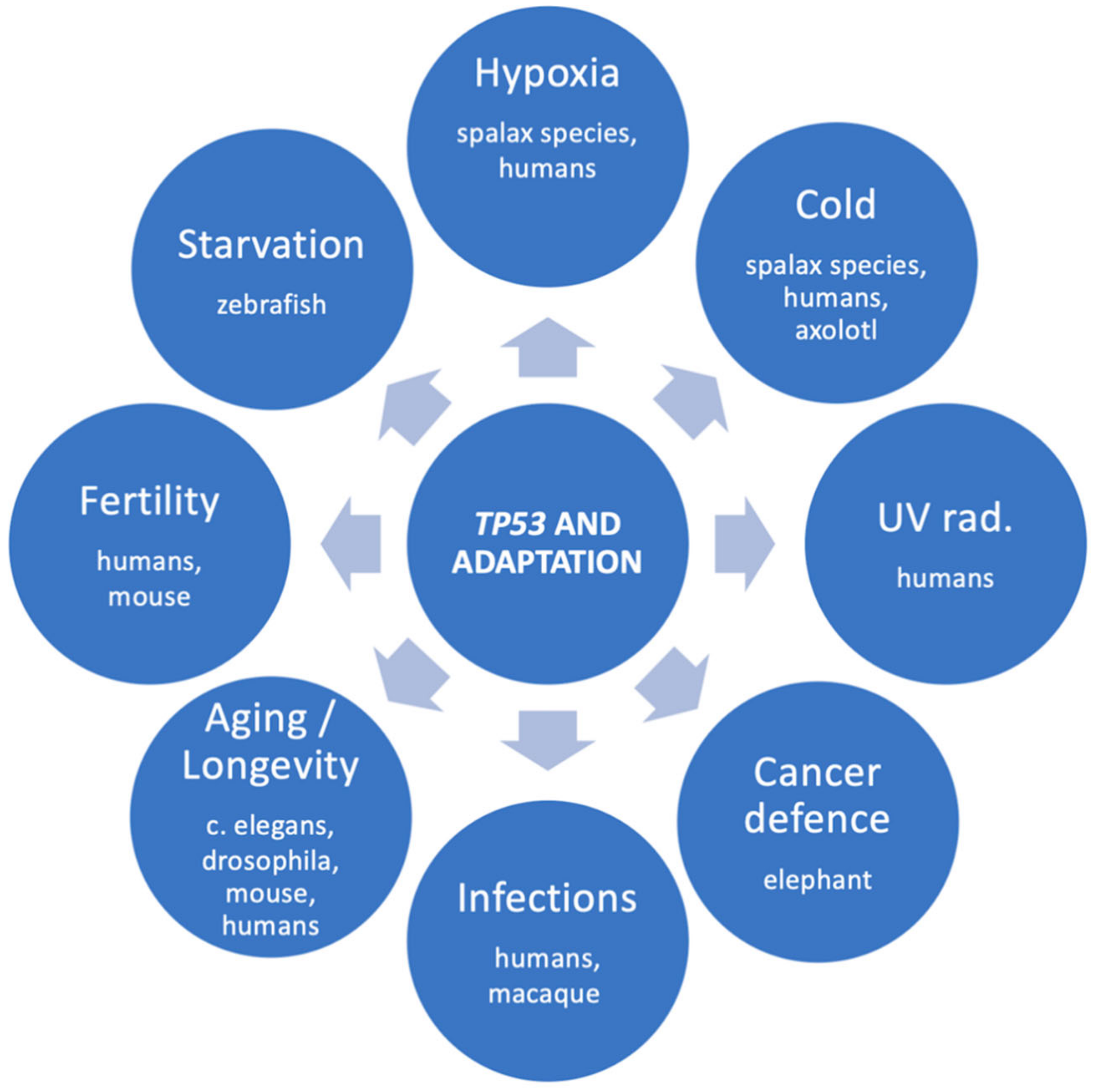
There is no doubt that TP53 promotes adaptation in many species. The adaptational role of TP53 is summarized in Figure 2. Sound examples include the TP53-R72P single amino-acid substitution in humans and the multiple TP53 copies in elephants. The obvious advantage of TP53 selection pressures is, of course, the defense against cancer. However, evidence shows that TP53 adaptive variants have been selected in multiple species for surviving under demanding conditions, such as cold, hypoxia, and probably viral infections, as well. Probably, there are many more adaptations to be discovered. High-throughput genetic methods will highly facilitate research of the next few years, and more intriguing aspects of the TP53 gene will be revealed.
Author Contributions
Conceptualization: K.V.; investigation: K.V. and N.G.; supervision: K.V.; writing-original draft: K.V. and N.G.; writing-review and editing: K.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Capuozzo, M.; Santorsola, M.; Bocchetti, M.; Perri, F.; Cascella, M.; Granata, V.; Celotto, V.; Gualillo, O.; Cossu, A.M.; Nasti, G.; et al. P53: From Fundamental Biology to Clinical Applications in Cancer. Biology 2022, 11, 1325. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant P53: It’s Not All One and the Same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Levine, A.J. P53: 800 Million Years of Evolution and 40 Years of Discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Levine, A.J. Spontaneous and Inherited TP53 Genetic Alterations. Oncogene 2021, 40, 5975–5983. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Cheok, C.F.; Brown, C.; Madhumalar, A.; Ghadessy, F.J.; Verma, C. Mdm2 and P53 Are Highly Conserved from Placozoans to Man. Cell Cycle 2010, 9, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zheng, T.; Wang, J. Regulation of Fertility by the P53 Family Members. Genes Cancer 2011, 2, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Beckman, G.; Birgander, R.; Sjalander, A.; Saha, N.; Holmberg, P.A.; Kivela, A.; Beckman, L. Is P53 Polymorphism Maintained by Natural Selection? Hum. Hered. 1994, 44, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.I.J.; Della Pietra, A.C.; George, D.L.; Murphy, M. The Codon 72 Polymorphic Variants of P53 Have Markedly Different Apoptotic Potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Hoyos, D.; Greenbaum, B.; Levine, A.J. The Genotypes and Phenotypes of Missense Mutations in the Proline Domain of the P53 Protein. Cell Death Differ. 2022, 29, 938–945. [Google Scholar] [CrossRef]
- Kang, H.J.; Feng, Z.; Sun, Y.; Atwal, G.; Murphy, M.E.; Rebbeck, T.R.; Rosenwaks, Z.; Levine, A.J.; Hu, W. Single-Nucleotide Polymorphisms in the P53 Pathway Regulate Fertility in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 9761–9766. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Feng, Z.; Teresky, A.K.; Levine, A.J. P53 Regulates Maternal Reproduction through LIF. Nature 2007, 450, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Lledo, B.; Turienzo, A.; Ortiz, J.A.; Morales, R.; Ten, J.; Llácer, J.; Bernabeu, R. Negative Effect of P72 Polymorphism on P53 Gene in IVF Outcome in Patients with Repeated Implantation Failure and Pregnancy Loss. J. Assist. Reprod. Genet. 2014, 31, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, X.; Wang, Z. Association between P53 Arg72Pro Polymorphism and Recurrent Pregnancy Loss: An Updated Systematic Review and Meta-Analysis. Reprod. Biomed. Online 2015, 31, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Tagliani-Ribeiro, A.; Paskulin, D.D.; Oliveira, M.; Zagonel-Oliveira, M.; Longo, D.; Ramallo, V.; Ashton-Prolla, P.; Saraiva-Pereira, M.L.; Fagundes, N.J.R.; Schuler-Faccini, L.; et al. High Twinning Rate in Cândido Godói: A New Role for P53 in Human Fertility. Hum. Reprod. 2012, 27, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ganeshan, L.; O’Neill, C. The Effect of Trp53 Gene-Dosage and Parent-of-Origin of Inheritance on Mouse Gamete and Embryo Function in Vitro. Biol. Reprod. 2012, 86, 1–6. [Google Scholar] [CrossRef]
- Byars, S.G.; Voskarides, K. Antagonistic Pleiotropy in Human Disease. J. Mol. Evol. 2020, 88, 12–25. [Google Scholar] [CrossRef]
- Bauer, J.H.; Poon, P.C.; Glatt-Deeley, H.; Abrams, J.M.; Helfand, S.L. Neuronal Expression of P53 Dominant-Negative Proteins in Adult Drosophila Melanogaster Extends Life Span. Curr. Biol. 2005, 15, 2063–2068. [Google Scholar] [CrossRef]
- Ingaramo, M.C.; Sánchez, J.A.; Dekanty, A. Regulation and Function of P53: A Perspective from Drosophila Studies. Mech. Dev. 2018, 154, 82–90. [Google Scholar] [CrossRef]
- Waskar, M.; Landis, G.N.; Shen, J.; Curtis, C.; Tozer, K.; Abdueva, D.; Skvortsov, D.; Tavaré, S.; Tower, J. Drosophila Melanogaster P53 Has Developmental Stage-Specific and Sex-Specific Effects on Adult Life Span Indicative of Sexual Antagonistic Pleiotropy. Aging 2009, 1, 903–936. [Google Scholar] [CrossRef]
- Arum, O.; Johnson, T.E. Reduced Expression of the Caenorhabditis Elegans P53 Ortholog Cep-1 Results in Increased Longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 951–959. [Google Scholar] [CrossRef] [Green Version]
- Yanase, S.; Suda, H.; Yasuda, K.; Ishii, N. Impaired P53/CEP-1 Is Associated with Lifespan Extension through an Age-Related Imbalance in the Energy Metabolism of C. Elegans. Genes Cells 2017, 22, 1004–1010. [Google Scholar] [CrossRef]
- Van Heemst, D.; Mooijaart, S.P.; Beekman, M.; Schreuder, J.; De Craen, A.J.M.; Brandt, B.W.; Eline Slagboom, P.; Westendorp, R.G.J. Variation in the Human TP53 Gene Affects Old Age Survival and Cancer Mortality. Exp. Gerontol. 2005, 40, 11–15. [Google Scholar] [CrossRef]
- Bojesen, S.E.; Nordestgaard, B.G. The Common Germline Arg72Pro Polymorphism of P53 and Increased Longevity in Humans. Cell Cycle 2008, 7, 158–163. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, L.; Yue, X.; Zhang, C.; Wang, J.; Li, J.; Sun, X.; Zhu, Y.; Feng, Z.; Hu, W. A Polymorphism in the Tumor Suppressor P53 Affects Aging and Longevity in Mouse Models. Elife 2018, 7, e34701. [Google Scholar] [CrossRef]
- Groß, S.; Immel, U.-D.; Klintschar, M.; Bartel, F. Germline Genetics of the P53 Pathway Affect Longevity in a Gender Specific Manner. Curr. Aging Sci. 2014, 7, 91–100. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. P53 Tetramerization: At the Center of the Dominant-Negative Effect of Mutant P53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Mehta, S.; Campbell, H.; Drummond, C.J.; Li, K.; Murray, K.; Slatter, T.; Bourdon, J.; Braithwaite, A.W. Adaptive Homeostasis and the P53 Isoform Network. EMBO Rep. 2021, 22, e53085. [Google Scholar] [CrossRef]
- Ashur-Fabian, O.; Avivi, A.; Trakhtenbrot, L.; Adamsky, K.; Cohen, M.; Kajakaro, G.; Joel, A.; Amariglio, N.; Nevo, E.; Rechavi, G. Evolution of P53 in Hypoxia-Stressed Spalax Mimics Human Tumor Mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 12236–12241. [Google Scholar] [CrossRef]
- Ellis, M.; Stern, O.; Ashur-Fabian, O. The Double Benefit of Spalax P53: Surviving Underground Hypoxia While Defying Lung Cancer Cells in Vitro via Autophagy and Caspase-Dependent Cell Death. Oncotarget 2016, 7, 63242–63251. [Google Scholar] [CrossRef]
- Shams, I.; Malik, A.; Manov, I.; Joel, A.; Band, M.; Avivi, A. Transcription Pattern of P53-Targeted DNA Repair Genes in the Hypoxia-Tolerant Subterranean Mole Rat Spalax. J. Mol. Biol. 2013, 425, 1111–1118. [Google Scholar] [CrossRef]
- Band, M.; Ashur-Fabian, O.; Avivi, A. The Expression of P53-Target Genes in the Hypoxia-Tolerant Subterranean Mole-Rat Is Hypoxia-Dependent and Similar to Expression Patterns in Solid Tumors. Cell Cycle 2010, 9, 3367–3372. [Google Scholar] [CrossRef]
- Fang, X.; Nevo, E.; Han, L.; Levanon, E.Y.; Zhao, J.; Avivi, A.; Larkin, D.; Jiang, X.; Feranchuk, S.; Zhu, Y.; et al. Genome-Wide Adaptive Complexes to Underground Stresses in Blind Mole Rats Spalax. Nat. Commun. 2014, 5, 3966. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.L.; Wang, M.Y.; Zhang, S.T.; Liua, Y.; Li, M.; Cao, Y.B.; Zu, H.Y.; Chen, X.C.; Wu, C.I.; et al. Codon 104 Variation of P53 Gene Provides Adaptive Apoptotic Responses to Extreme Environments in Mammals of the Tibet Plateau. Proc. Natl. Acad. Sci. USA 2013, 110, 20639–20644. [Google Scholar] [CrossRef]
- Kimura, K.; Shinmura, K.; Hasegawa, T.; Beppu, Y.; Yokoyama, R.; Yokota, J. Germline P53 Mutation in a Patient with Multiple Primary Cancers. Jpn. J. Clin. Oncol. 2001, 31, 349–351. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, J.W.; Yang, Z.; Cao, Y.B.; Ren, J.L.; Ben-Abu, Y.; Li, K.; Chen, X.Q.; Du, J.Z.; Nevo, E. Adaptive Methylation Regulation of P53 Pathway in Sympatric Speciation of Blind Mole Rats, Spalax. Proc. Natl. Acad. Sci. USA 2016, 113, 2146–2151. [Google Scholar] [CrossRef]
- Villiard, É.; Brinkmann, H.; Moiseeva, O.; Mallette, F.A.; Ferbeyre, G.; Roy, S. Urodele P53 Tolerates Amino Acid Changes Found in P53 Variants Linked to Human Cancer. BMC Evol. Biol. 2007, 7, 180. [Google Scholar] [CrossRef]
- Jacovas, V.C.; Couto-Silva, C.M.; Nunes, K.; Lemes, R.B.; de Oliveira, M.Z.; Salzano, F.M.; Bortolini, M.C.; Hünemeier, T. Selection Scan Reveals Three New Loci Related to High Altitude Adaptation in Native Andeans. Sci. Rep. 2018, 8, 12733. [Google Scholar] [CrossRef]
- Jacovas, V.C.; Rovaris, D.L.; Peréz, O.; De Azevedo, S.; Macedo, G.S.; Sandoval, J.R.; Salazar-Granara, A.; Villena, M.; Dugoujon, J.M.; Bisso-Machado, R.; et al. Genetic Variations in the TP53 Pathway in Native Americans Strongly Suggest Adaptation to the High Altitudes of the Andes. PLoS ONE 2015, 10, e0137823. [Google Scholar] [CrossRef]
- Shi, H.; Tan, S.-J.; Zhong, H.; Hu, W.; Levine, A.; Xiao, C.-J.; Peng, Y.; Qi, X.-B.; Shou, W.-H.; Ma, R.-L.Z.; et al. Winter Temperature and UV Are Tightly Linked to Genetic Changes in the P53 Tumor Suppressor Pathway in Eastern Asia. Am. J. Hum. Genet. 2009, 84, 534–541. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Voskarides, K.; Koutsofti, C.; Pozova, M. TP53 Mutant Versus Wild-Type Zebrafish Larvae Under Starvation Stress: Larvae Can Live Up to 17 Days Post-Fertilization Without Food. Zebrafish 2022, 19, 49–55. [Google Scholar] [CrossRef]
- Thomas, M.; David, P.; Banks, L. The Role of the E6-P53 Interaction in the Molecular Pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef]
- Long, T.; Burk, R.D.; Chan, P.K.S.; Chen, Z. Non-Human Primate Papillomavirus E6-Mediated P53 Degradation Reveals Ancient Evolutionary Adaptation of Carcinogenic Phenotype to Host Niche. PLoS Pathog. 2022, 18, e1010444. [Google Scholar] [CrossRef]
- Tomasetti, C. Mutated Clones Are the New Normal: Measuring and Understanding the Dynamics of Clonal Cell Populations Is Key for Cancer Prevention. Science 2019, 364, 938–939. [Google Scholar] [CrossRef]
- Luijts, T.; Elliott, K.; Siaw, J.T.; Van de Velde, J.; Beyls, E.; Claeys, A.; Lammens, T.; Larsson, E.; Willaert, W.; Vral, A.; et al. A Clinically Annotated Post-Mortem Approach to Study Multi-Organ Somatic Mutational Clonality in Normal Tissues. Sci. Rep. 2022, 12, 10322. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Colom, B.; Alcolea, M.P.; Piedrafita, G.; Hall, M.W.J.; Wabik, A.; Dentro, S.C.; Fowler, J.C.; Herms, A.; King, C.; Ong, S.H.; et al. Spatial Competition Shapes the Dynamic Mutational Landscape of Normal Esophageal Epithelium. Nat. Genet. 2020, 52, 604–614. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-Related Remodelling of Oesophageal Epithelia by Mutated Cancer Drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The Landscape of Somatic Mutation in Normal Colorectal Epithelial Cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco Smoking and Somatic Mutations in Human Bronchial Epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef]
- Li, R.; Du, Y.; Chen, Z.; Xu, D.; Lin, T.; Jin, S.; Wang, G.; Liu, Z.; Lu, M.; Chen, X.; et al. Macroscopic Somatic Clonal Expansion in Morphologically Normal Human Urothelium. Science 2020, 370, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Herms, A.; Hall, M.W.J.; Dentro, S.C.; King, C.; Sood, R.K.; Alcolea, M.P.; Piedrafita, G.; Fernandez-Antoran, D.; Ong, S.H.; et al. Mutant Clones in Normal Epithelium Outcompete and Eliminate Emerging Tumours. Nature 2021, 598, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and P16(INK4a). Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boddy, A.M.; Maley, C.C. Peto’s Paradox: How Has Evolution Solved the Problem of Cancer Prevention? BMC Biol. 2017, 15, 60. [Google Scholar] [CrossRef]
- Peto, H.; Roe, F.J.C.; Lee, P.N.; Levy, L.; Clack, J. Cancer and Ageing in Mice and Men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef]
- Nunney, L. The Real War on Cancer: The Evolutionary Dynamics of Cancer Suppression. Evol. Appl. 2013, 6, 11–19. [Google Scholar] [CrossRef]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA J. Am. Med. Assoc. 2015, 314, 1850–1860. [Google Scholar] [CrossRef]
- García-Cao, I.; García-Cao, M.; Martín-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super P53” Mice Exhibit Enhanced DNA Damage Response, Are Tumor Resistant and Age Normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef] [PubMed]
- Sulak, M.; Fong, L.; Mika, K.; Chigurupati, S.; Yon, L.; Mongan, N.P.; Emes, R.D.; Lynch, V.J. TP53 Copy Number Expansion Is Associated with the Evolution of Increased Body Size and an Enhanced DNA Damage Response in Elephants. eLife 2016, 5, e11994. [Google Scholar] [CrossRef] [PubMed]
- Padariya, M.; Jooste, M.L.; Hupp, T.; Fåhraeus, R.; Vojtesek, B.; Vollrath, F.; Kalathiya, U.; Karakostis, K. The Elephant Evolved P53 Isoforms That Escape MDM2-Mediated Repression and Cancer. Mol. Biol. Evol. 2022, 39, msac149. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenetic gain/loss tree of TP53 gene (created in www.ensembl.com on 17 December 2022). The tree shows the number of TP53 paralogues/copies for each species or taxon. Red lines mark significant expansion of the TP53 gene copies.
Figure 1.
Phylogenetic gain/loss tree of TP53 gene (created in www.ensembl.com on 17 December 2022). The tree shows the number of TP53 paralogues/copies for each species or taxon. Red lines mark significant expansion of the TP53 gene copies.

Figure 2.
The multiple adaptational roles of TP53 and the species that evidence was found for each role.
Figure 2.
The multiple adaptational roles of TP53 and the species that evidence was found for each role.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Voskarides, K.; Giannopoulou, N. The Role of TP53 in Adaptation and Evolution. Cells 2023, 12, 512. https://doi.org/10.3390/cells12030512
AMA Style
Voskarides K, Giannopoulou N. The Role of TP53 in Adaptation and Evolution. Cells. 2023; 12(3):512. https://doi.org/10.3390/cells12030512
Chicago/Turabian StyleVoskarides, Konstantinos, and Nefeli Giannopoulou. 2023. "The Role of TP53 in Adaptation and Evolution" Cells 12, no. 3: 512. https://doi.org/10.3390/cells12030512
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.