


Immunogenomic Biomarkers and Validation in Lynch Syndrome
 ,
,
Abstract
:1. Introduction
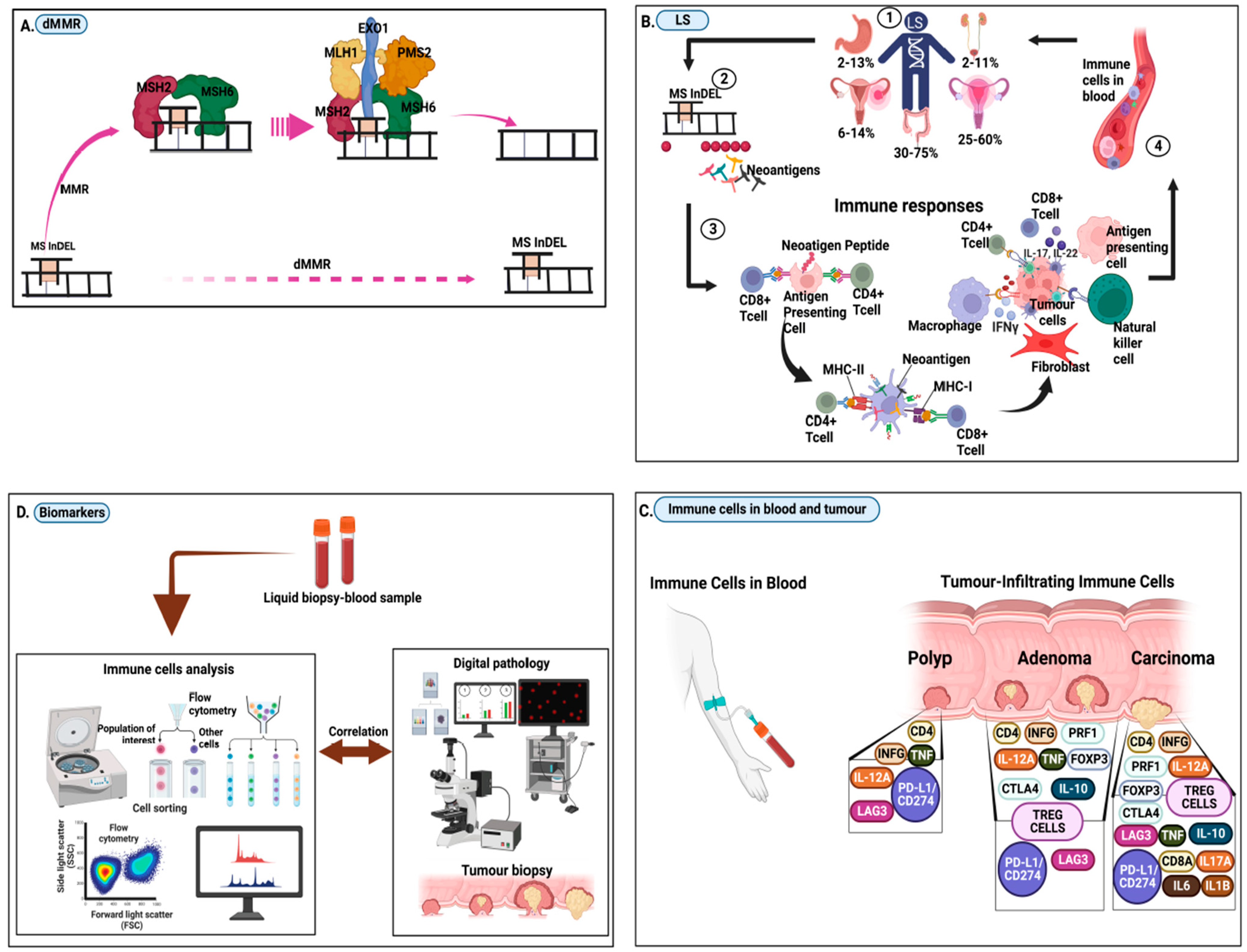
2. Frameshift Neopeptides, Neoantigens, and the Immune Responses in LS
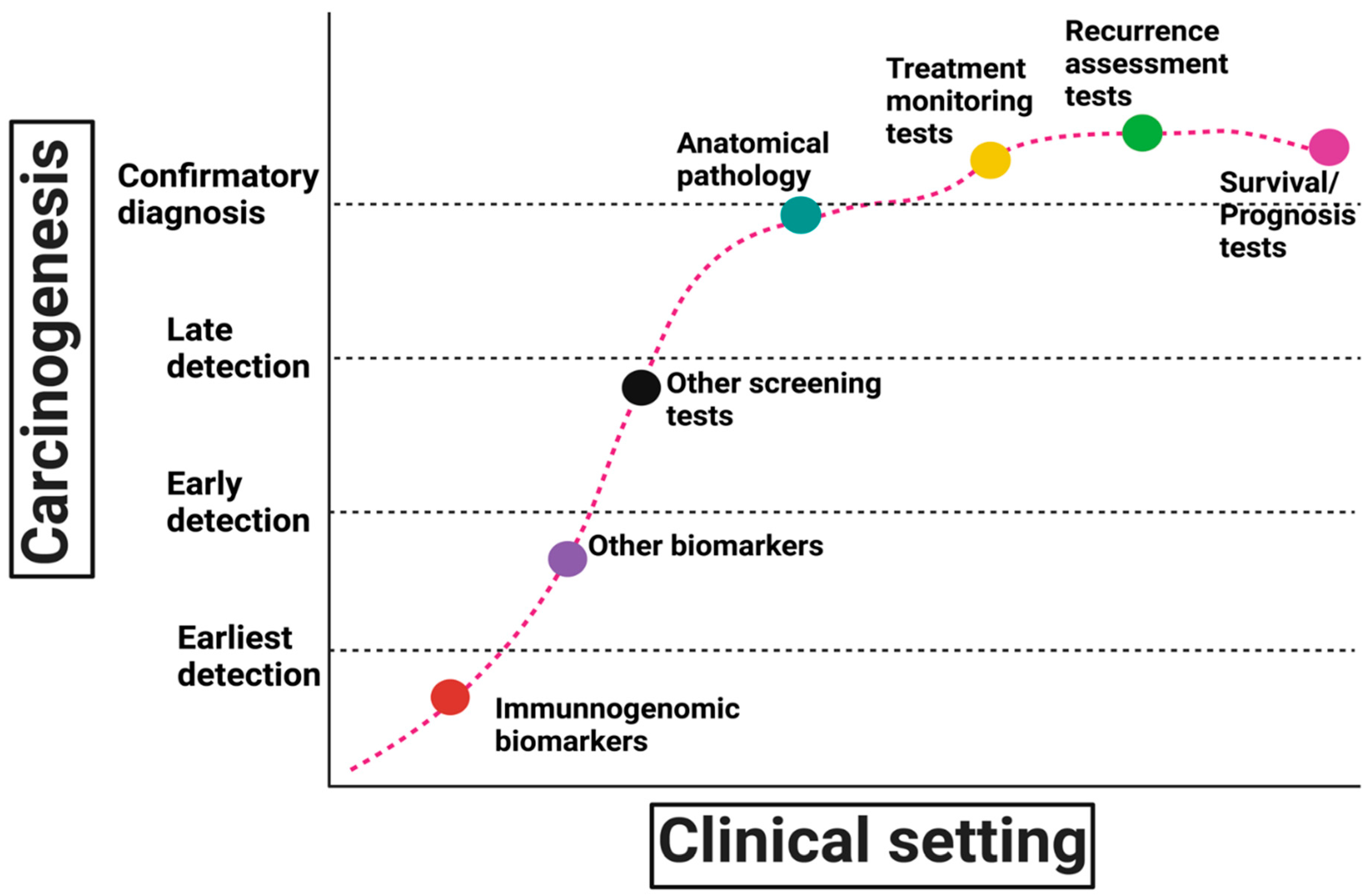
3. Research Questions in Lynch Syndrome Immunogenomic Biomarkers
- (i)
- How can the measurable immunogenomic components in LSVH that occur as a result of mutations in the mismatch-repair genes be characterised?
- Rationale:
- (ii)
- Could immunogenomic biomarker profiles serve endophenotypically as potential biomarkers to reflect neoplastic changes (from early-stage to invasive and metastatic cancer) in LSVH?
- Rationale:
- (iii)
- Can immunogenomic biomarker profiles serve to prognosticate, i.e., predict disease-free survival and overall survival for LSVH carrying the same or different novel PV?
- Rationale:

{kind=link}
{kind=link}
| 1. IMMUNOGENOMIC BIOMARKERS | 2. OTHER BIOMARKERS |
|---|---|
| Panel 1 biomarker profiles (for polyp) CD4,INFG, LAG3,PDL1,/CD274, IL12A, TNF [34] | A. Specific for colorectal cancer (i) Faecal occult blood testing (ii) Stool DNA, miRNA [24,70] (iii) Faecal immunological test (FIT) [71] (iv) Faecal bacteria (v) Gut microbiota signatures [72] |
| Panel 2 biomarker profiles (for adenoma) PRF1,FOXP3,CTLA4, IL-10,TREG CELLS [34] | B. Colorectal and non-colorectal cancers (i) DNA, RNA, cfDNA, ctDNA, cfRNA, mRNA, microRNA, IncRNA (ii) Circulating tumour cells (iii) CA 125 Blood test [73] (iv) Methylation tests [23] (v) Growth factors tests (vi) Tissue tests (vii) Proteins and Glycoproteins tests [74] (viii) Tissue tests (ix) Volatile organic compounds (VOC) [75] (x) Immune-Inflammation index [68] (xi) Prostate cancer antigen 3 test (PCA3) [76] (xii) Genomic Prostate Score [77] |
| Panel 3 biomarker profiles (for carcinoma) CD8A, IL17A, IL1B, IL6 [34] |
4. Validation of Immunogenomic Biomarkers
4.1. Pre-Analytical Validation
- i.
- Pre-assessment of biomarkers and ensuring an expedient approach to assay development;
- ii.
- Consider quality assurance and quality control procedures for blood-based assays for each specific biomarker panel;
- iii.
- Maintain optimal pre-analytical processing procedures and SOPs for control of specific biomarkers;
- iv.
- During analytical validation, it is advisable to use procedures that include strict quality assurance, reproducibility protocols, and control procedures;
- v.
- Whenever possible, reagents and assay controls (positive and negative controls) must be included in the interpretation of assay results;
- vi.
- Biostatistical and computational approaches to quantification and interpretation of data must be considered. In addition, the development of algorithms for multiplex signatures based on phenotypic, functional, and genomic data must be considered;
- vii.
- An integrated bioinformatics approach needs to be considered for the integration of complex high-throughput immunogenomic data consisting of multiple components;
- viii.
- The use of reference standards and/or coordination efforts between centralized laboratories (proficiency panels) is recommended to assess the robustness of semi-quantitative methods and to enable analytical and clinical validation of biomarkers.
4.2. Outcome Measures
- A.
- Primary outcome measureTo test the sensitivity of a blood-based panel of immunogenomic biomarkers against colonoscopy and other cancer screening methods. A consensus panel based on blood samples will be able to detect colorectal adenocarcinoma with a significant improvement in sensitivity and specificity [80,84]. However, the persistence of immunogenomic biomarkers in the blood of already diagnosed and treated patients may affect the specificity of the biomarkers. This is because they may remain at high levels even after treatment for cancer or precancerous lesions.
- B.
- Secondary Outcome Measures
- i.
- To test the specificity of blood-based immunogenomic biomarkers compared with pathology tests [80], using blood-based panels to detect colorectal adenocarcinoma with the same or higher sensitivity as pathology tests. The hypothesis is that they have a significantly higher specificity than 0.55 with an expected specificity greater than 0.70.
- ii.
- To analyse the sensitivity and specificity of a combined panel of blood-based immunogenomic biomarkers for CRC detection [80,84]. To test the hypothesis that combining the blood-based panel with the tissue-based panel will improve the detection of colorectal adenocarcinoma: with a sensitivity greater than 0.98, it will have a specificity of 0.55 or greater.
Using the area under the receiver operating characteristic (ROC) curves and logistic regression models, models that have high sensitivity, specificity, positive predictive value, and negative predictive value for advanced neoplasms detection compared to healthy individuals were found [85]. By multiplex flow cytometry, which examines a range of lymphocyte markers, phenotypic analysis of T cells can provide information about their activation status. A baseline signature of immune cells and pro-inflammatory markers with a higher baseline frequency of CD4+CD25+FoxP3+ Tregs should be identified. For this immunogenomic biomarker signature to be used in routine clinical practice, it needs to be analytically and clinically validated (including a panel of markers required to analyse and enumerate cells).
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular pathology of Lynch syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef]
- Tibiletti, M.G.; Carnevali, I.; Calò, V.; Cini, G.; Cordisco, E.L.; Remo, A.; Urso, E.; Oliani, C.; Ranzani, G.N. Universal testing for MSI/MMR status in colorectal and endometrial cancers to identify Lynch syndrome cases: State of the art in Italy and consensus recommendations from the Italian Association for the Study of Familial Gastrointestinal Tumors (A.I.F.E.G.). Eur. J. Cancer Prev. 2022, 31, 44–49. [Google Scholar] [CrossRef]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.; Shaw, T.; Heinen, C.; Hitchins, M. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, G.; Wu, W. Recent advances in Lynch syndrome. Exp. Hematol. Oncol. 2021, 10, 37. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.; Balaguer, F.; Lindor, N.; De La Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch Syndrome Among Patients with Colorectal Cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Gupta, S.; Burke, C.A.; Axell, L.; Chen, L.-M.; Chung, D.C.; Clayback, K.M.; Dallas, S.; Felder, S.; Gbolahan, O.; et al. NCCN Guidelines® Insights: Genetic/Familial High-Risk Assessment: Colorectal.; Version 1.2021: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2021, 19, 1122–1132. [Google Scholar] [CrossRef]
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Stupart, D.A.; Goldberg, P.; Algar, U.; Ramesar, R. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Color. Dis. 2009, 11, 126–130. [Google Scholar] [CrossRef]
- Wentink, M.Q.; Räkers, M.; Stupart, D.A.; Algar, U.; Ramesar, R.; Goldberg, P. Incidence and histological features of colorectal cancer in the Northern Cape Province, South Africa. South Afr. J. Surg. 2010, 48, 109–113. [Google Scholar]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Bruwer, Z.; Futter, M.; Ramesar, R. A Mobile Colonoscopic Unit for Lynch Syndrome: Trends in Surveillance Uptake and Patient Experiences of Screening in a Developing Country. J. Genet. Couns. 2013, 22, 125–137. [Google Scholar] [CrossRef]
- Adler, A.; Geiger, S.; Keil, A.; Bias, H.; Schatz, P.; Devos, T.; Dhein, J.; Zimmermann, M.; Tauber, R.; Wiedenmann, B. Improving compliance to colorectal cancer screening using blood and stool based tests in patients refusing screening colonoscopy in Germany. BMC Gastroenterol. 2014, 14, 183. [Google Scholar] [CrossRef]
- Mittendorf, K.F.; Knerr, S.; Kauffman, T.L.; Lindberg, N.M.; Anderson, K.P.; Feigelson, H.S.; Gilmore, M.J.; Hunter, J.E.; Joseph, G.; Kraft, S.A.; et al. Systemic Barriers to Risk-Reducing Interventions for Hereditary Cancer Syndromes: Implications for Health Care Inequities. JCO Precis. Oncol. 2021, 5, 1709–1718. [Google Scholar] [CrossRef]
- Patel, S.; Ahnen, D.J.; Kinney, A.; Horick, N.; Finkelstein, D.M.; Hill, D.A.; Lindor, N.M.; MaCrae, F.; Lowery, J.T. Knowledge and Uptake of Genetic Counseling and Colonoscopic Screening Among Individuals at Increased Risk for Lynch Syndrome and their Endoscopists from the Family Health Promotion Project. Am. J. Gastroenterol. 2016, 111, 285–293. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef]
- Lorans, M.; Dow, E.; Macrae, F.A.; Winship, I.M.; Buchanan, D.D. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin. Color. Cancer 2018, 17, e293–e305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, A.K.; Dowty, J.G.; Reece, J.C.; Lee, G.; Templeton, A.S.; Plazzer, J.-P.; Buchanan, D.D.; Akagi, K.; Aksoy, S.; Alonso, A.; et al. Variation in the risk of colorectal cancer in families with Lynch syndrome: A retrospective cohort study. Lancet Oncol. 2021, 22, 1014–1022. [Google Scholar] [CrossRef]
- Loomans-Kropp, H.A.; Song, Y.; Gala, M.; Parikh, A.R.; Van Seventer, E.E.; Alvarez, R.; Hitchins, M.P.; Shoemaker, R.H.; Umar, A. Methylated Septin9 (mSEPT9): A Promising Blood-Based Biomarker for the Detection and Screening of Early-Onset Colorectal Cancer. Cancer Res. Commun. 2022, 2, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, A.; Vlachostergios, P.; Oikonomou, K.G.; Moshenyat, Y. Fecal DNA testing for colorectal cancer screening: Molecular targets and perspectives. World J. Gastrointest. Oncol. 2015, 7, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Zygulska, A.L.; Pierzchalski, P. Novel Diagnostic Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 852. [Google Scholar] [CrossRef]
- Kamel, F.; Eltarhoni, K.; Nisar, P.; Soloviev, M. Colorectal Cancer Diagnosis: The Obstacles We Face in Determining a Non-Invasive Test and Current Advances in Biomarker Detection. Cancers 2022, 14, 1889. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Win, A.K.; Scott, R.J. Genetic and Environmental Modifiers of Cancer Risk in Lynch Syndrome, in Hereditary Colorectal Cancer: Genetic Basis and Clinical Implications; Valle, L., Gruber, S.B., Capellá, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 67–89. [Google Scholar]
- Kupfer, S.S. Broadening our Understanding of the Immune Landscape in Lynch Syndrome. Gastroenterology 2022, 162, 1024–1025. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: A report from the prospective Lynch syndrome database. Gut 2017, 66, 1657–1664. [Google Scholar] [CrossRef]
- Ballester, V. A step closer to a personalised approach for Lynch syndrome. Lancet Oncol. 2021, 22, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Sanchez, A.; Grossman, M.; Yeung, K.; Sei, S.S.; Lipkin, S.; Kloor, M. Vaccines for immunoprevention of DNA mismatch repair deficient cancers. J. Immunother. Cancer 2022, 10, e004416. [Google Scholar] [CrossRef] [PubMed]
- Pastor, D.M.; Schlom, J. Immunology of Lynch Syndrome. Curr. Oncol. Rep. 2021, 23, 96. [Google Scholar] [CrossRef]
- Kurz, C.; Hakimi, M.; Kloor, M.; Grond-Ginsbach, C.; Gross-Weissmann, M.-L.; Böckler, D.; Doeberitz, M.V.K.; Dihlmann, S. Coding Microsatellite Frameshift Mutations Accumulate in Atherosclerotic Carotid Artery Lesions: Evaluation of 26 Cases and Literature Review. Mol. Med. 2015, 21, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef]
- Kloor, M.; Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.-R.; Al-Batran, S.-E.; Tariverdian, M.; Jäger, E.; Doeberitz, M.V.K. A Frameshift Peptide Neoantigen-Based Vaccine for Mismatch Repair-Deficient Cancers: A Phase I/IIa Clinical Trial. Clin. Cancer Res. 2020, 26, 4503–4510. [Google Scholar] [CrossRef]
- Doeberitz, M.V.K.; Kloor, M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam. Cancer 2013, 12, 307–312. [Google Scholar] [CrossRef]
- Bauer, K.; Nelius, N.; Reuschenbach, M.; Koch, M.; Weitz, J.; Steinert, G.; Kopitz, J.; Beckhove, P.; Tariverdian, M.; Doeberitz, M.V.K.; et al. T cell responses against microsatellite instability-induced frameshift peptides and influence of regulatory T cells in colorectal cancer. Cancer Immunol. Immunother. 2013, 62, 27–37. [Google Scholar] [CrossRef]
- Bohaumilitzky, L.; Kluck, K.; Hüneburg, R.; Gallon, R.; Nattermann, J.; Kirchner, M.; Kristiansen, G.; Hommerding, O.; Pfuderer, P.L.; Wagner, L.; et al. The Different Immune Profiles of Normal Colonic Mucosa in Cancer-Free Lynch Syndrome Carriers and Lynch Syndrome Colorectal Cancer Patients. Gastroenterology 2022, 162, 907–919.e10. [Google Scholar] [CrossRef]
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A., Jr. Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. Oncologist 2016, 21, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Syngal, S.; Yurgelun, M.B. Recent advances in Lynch syndrome. Fam. Cancer 2019, 18, 211–219. [Google Scholar] [CrossRef]
- Sedhom, R.; Antonarakis, E.S. Clinical implications of mismatch repair deficiency in prostate cancer. Futur. Oncol. 2019, 15, 2395–2411. [Google Scholar] [CrossRef]
- Willis, J.A.; Reyes-Uribe, L.; Chang, K.; Lipkin, S.M.; Vilar, E. Immune Activation in Mismatch Repair–Deficient Carcinogenesis: More Than Just Mutational Rate. Clin. Cancer Res. 2020, 26, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Roudko, V.; Bozkus, C.C.; Greenbaum, B.; Lucas, A.; Samstein, R.; Bhardwaj, N. Lynch Syndrome and MSI-H Cancers: From Mechanisms to “Off-The-Shelf” Cancer Vaccines. Front. Immunol. 2021, 12, 757804. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A., III; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. Cancer and the Immune System: Basic Concepts and Targets for Intervention. Semin. Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015, 75, 3446–3455. [Google Scholar] [CrossRef]
- Maby, P.; Galon, J.; Latouche, J.-B. Frameshift mutations, neoantigens and tumor-specific CD8+T cells in microsatellite unstable colorectal cancers. Oncoimmunology 2016, 5, e1115943. [Google Scholar] [CrossRef]
- Ye, B.; Stary, C.M.; Gao, Q.; Wang, Q.; Zeng, Z.; Jian, Z.; Gu, L.; Xiong, X. Genetically Modified T-Cell-Based Adoptive Immunotherapy in Hematological Malignancies. J. Immunol. Res. 2017, 2017, 5210459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Bakarurraini, N.A.A.R.; Ab Mutalib, N.S.; Jamal, R.; Abu, N. The Landscape of Tumor-Specific Antigens in Colorectal Cancer. Vaccines 2020, 8, 371. [Google Scholar] [CrossRef]
- Chambuso, R.; Kaambo, E.; Rebello, G.; Ramesar, R. Correspondence on “Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database” by Dominguez-Valentin et al. Anesthesia Analg. 2022, 24, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Prospective Lynch Syndrome Database (PLSD), in European Hereditary Tumor Group (EHTG), Centres, E., Ed. 2012. Available online: http://www.plsd.eu (accessed on 12 October 2022).
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Na Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Olsson, N.; Heberling, M.L.; Zhang, L.; Jhunjhunwala, S.; Phung, Q.T.; Lin, S.; Anania, V.G.; Lill, J.R.; Elias, J.E. An Integrated Genomic, Proteomic, and Immunopeptidomic Approach to Discover Treatment-Induced Neoantigens. Front. Immunol. 2021, 12, 662443. [Google Scholar] [CrossRef]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Stone, W.L.; Basit, H.; Burns, B. Pathology, Inflammation; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Woerner, S.M.; Yuan, Y.P.; Benner, A.; Korff, S.; von Knebel Doeberitz, M.; Bork, P. SelTarbase, a database of human mononucleotide-microsatellite mutations and their potential impact to tumorigenesis and immunology. Nucleic Acids Res. 2010, 38, D682–D689. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Hernandez Sanchez, A.; Urban, K.; Draxlbauer, M.; et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020, 11, 4740. [Google Scholar] [CrossRef]
- de Miranda, N.F.; Goudkade, D.; Jordanova, E.S.; Tops, C.M.; Hes, F.J.; Vasen, H.F.; van Wezel, T.; Morreau, H. Infiltration of Lynch Colorectal Cancers by Activated Immune Cells Associates with Early Staging of the Primary Tumor and Absence of Lymph Node Metastases. Clin. Cancer Res. 2012, 18, 1237–1245. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune Responses to Cancer: Are They Potential Biomarkers of Prognosis? Front. Oncol. 2013, 3, 107. [Google Scholar] [CrossRef]
- Ballman, K.V. Biomarker: Predictive or Prognostic? J. Clin. Oncol. 2015, 33, 3968–3971. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gu, L.; Chen, Y.; Chong, Y.; Wang, X.; Guo, P.; He, D. Systemic immune-inflammation index is a promising non-invasive biomarker for predicting the survival of urinary system cancers: A systematic review and meta-analysis. Ann. Med. 2021, 53, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Ribeiro, I.; Jorge, J.; Gonçalves, A.; Sarmento-Ribeiro, A.; Melo, J.; Carreira, I. Liquid Biopsies: Applications for Cancer Diagnosis and Monitoring. Genes 2021, 12, 349. [Google Scholar] [CrossRef] [PubMed]
- Le Pimpec, F.; Moutel, G.; Piette, C.; Lièvre, A.; Bretagne, J.-F. Fecal immunological blood test is more appealing than the guaiac-based test for colorectal cancer screening. Dig. Liver Dis. 2017, 49, 1267–1272. [Google Scholar] [CrossRef]
- Konishi, Y.; Okumura, S.; Matsumoto, T.; Itatani, Y.; Nishiyama, T.; Okazaki, Y.; Shibutani, M.; Ohtani, N.; Nagahara, H.; Obama, K.; et al. Development and evaluation of a colorectal cancer screening method using machine learning-based gut microbiota analysis. Cancer Med. 2022, 11, 3194–3206. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. CA-125 blood test in early detection of ovarian cancer. Lancet Oncol. 2015, 16, e269. [Google Scholar] [CrossRef]
- Nikolaou, S.; Qiu, S.; Fiorentino, F.; Rasheed, S.; Tekkis, P.; Kontovounisios, C. Systematic review of blood diagnostic markers in colorectal cancer. Tech. Coloproctol. 2018, 22, 481–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Tao, J.; Li, J.; Tao, S. Volatile organic compounds analysis as a potential novel screening tool for colorectal cancer: A systematic review and meta-analysis. Medicine 2020, 99, e20937. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.F.; Eggener, S.E. Prostate Cancer and the Evolving Role of Biomarkers in Screening and Diagnosis. Radiol. Clin. N. Am. 2018, 56, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, Z.; Cowan, J.E.; Westphalen, A.C.; Cooperberg, M.R.; Chan, J.M.; Zhao, S.; Shinohara, K.; Carroll, P.R. Genomic Prostate Score, PI-RADS™ version 2 and Progression in Men with Prostate Cancer on Active Surveillance. J. Urol. 2019, 201, 300–307. [Google Scholar] [CrossRef]
- Dobbin, K.K.; Cesano, A.; Alvarez, J.; Hawtin, R.; Janetzki, S.; Kirsch, I.; Masucci, G.V.; Robbins, P.B.; Selvan, S.R.; Streicher, H.Z.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume II—Clinical validation and regulatory considerations. J. Immunother. Cancer 2016, 4, 77. [Google Scholar] [CrossRef]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Mazzone, P.J.; Sears, C.R.; Arenberg, D.A.; Gaga, M.; Gould, M.K.; Massion, P.P.; Nair, V.S.; Powell, C.A.; Silvestri, G.A.; Vachani, A.; et al. Evaluating Molecular Biomarkers for the Early Detection of Lung Cancer: When Is a Biomarker Ready for Clinical Use? An Official American Thoracic Society Policy Statement. Am. J. Respir. Crit. Care Med. 2017, 196, e15–e29. [Google Scholar] [CrossRef]
- Hayes, D.F. Biomarker validation and testing. Mol. Oncol. 2015, 9, 960–966. [Google Scholar] [CrossRef]
- Masucci, G.V.; Cesano, A.; Hawtin, R.; Janetzki, S.; Zhang, J.; Kirsch, I.; Dobbin, K.K.; Alvarez, J.; Robbins, P.B.; Selvan, S.R.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume I—Pre-analytical and analytical validation. J. Immunother. Cancer 2016, 4, 76. [Google Scholar] [CrossRef]
- Godfrey, A.; Vandendriessche, B.; Bakker, J.P.; Fitzer-Attas, C.; Gujar, N.; Hobbs, M.; Liu, Q.; Northcott, C.A.; Parks, V.; Wood, W.A.; et al. Fit-for-Purpose Biometric Monitoring Technologies: Leveraging the Laboratory Biomarker Experience. Clin. Transl. Sci. 2021, 14, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-J.; Chang, Y.-C.I.; Hsueh, H.-M. Biomarker selection for medical diagnosis using the partial area under the ROC curve. BMC Res. Notes 2014, 7, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chambuso, R.; Mthembu, M.; Kaambo, E.; Robertson, B.; Ramesar, R. Immunogenomic Biomarkers and Validation in Lynch Syndrome. Cells 2023, 12, 491. https://doi.org/10.3390/cells12030491
Chambuso R, Mthembu M, Kaambo E, Robertson B, Ramesar R. Immunogenomic Biomarkers and Validation in Lynch Syndrome. Cells. 2023; 12(3):491. https://doi.org/10.3390/cells12030491
Chicago/Turabian StyleChambuso, Ramadhani, Mbali Mthembu, Eveline Kaambo, Barbara Robertson, and Raj Ramesar. 2023. "Immunogenomic Biomarkers and Validation in Lynch Syndrome" Cells 12, no. 3: 491. https://doi.org/10.3390/cells12030491