
RIPK1 Regulates Microglial Activation in Lipopolysaccharide-Induced Neuroinflammation and MPTP-Induced Parkinson’s Disease Mouse Models
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. BV2 Microglial Cell Culture
2.3. Reagents and Antibodies
2.4. Measurement of Nitrite, Cytokine, and Intracellular Reactive Oxygen Species (ROS) Levels
2.5. Cytotoxicity Assay (LDH Assay)
2.6. Western Blot Analysis
2.7. Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
2.8. Electrophoretic Mobility Shift Assay (EMSA)
2.9. Transient Transfection and Luciferase Assay
2.10. Drug Administration
2.11. Brain Tissue Preparation
2.12. Immunohistochemistry and Immunofluorescence Analysis
2.13. Statistical Analysis
3. Results
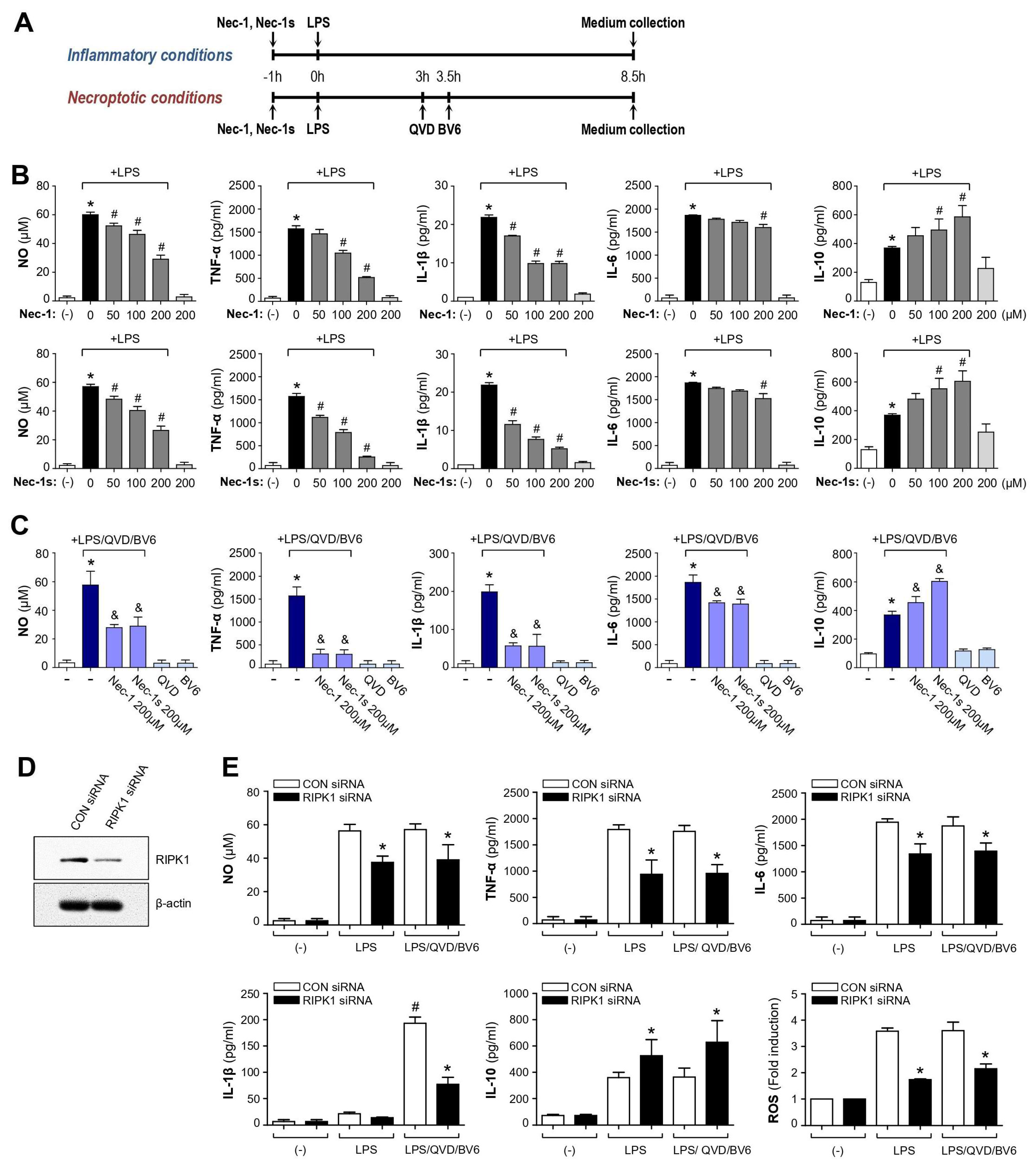
3.1. Nec-1 and Nec-1s Showed Anti-Inflammatory Effects in LPS or LPS/QVD/BV6-Stimulated BV2 Microglial Cells
3.2. Knockdown of RIPK1 Recapitulated the Anti-Inflammatory Effect of Nec-1/Nec-1s in LPS- or LPS/QVD/BV6-Stimulated BV2 Cells
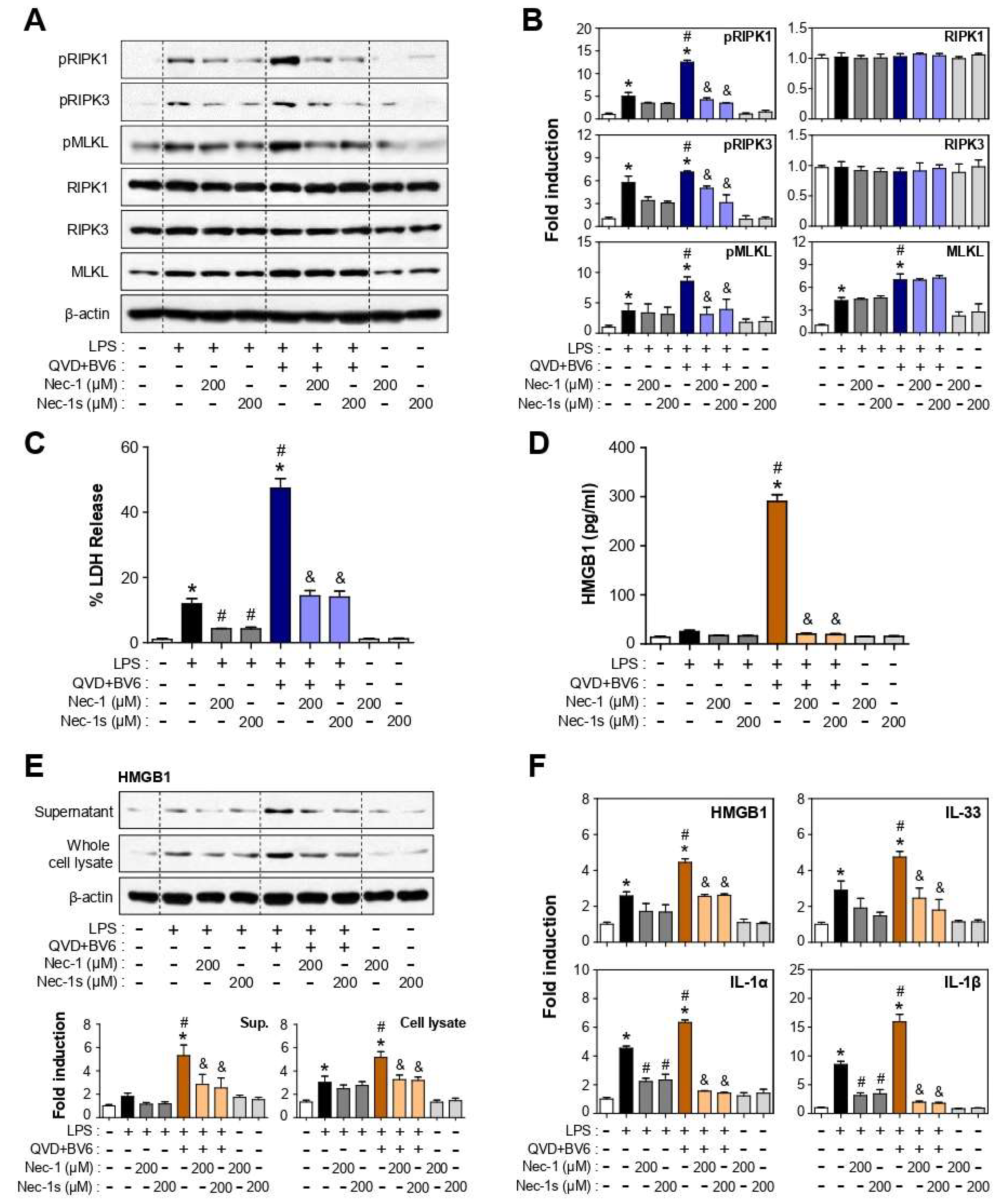
3.3. Nec-1 and Nec-1s Inhibited RIPK1-RIPK3-MLKL Phosphorylation and Cell Death in LPS or LPS/QVD/BV6-Stimulated BV2 Cells
3.4. Nec-1/Nec-1s Exerted Anti-Inflammatory Effects by Modulating AMPK, PI3K/Akt, MAPKs, and NF-κB Signaling Pathways in LPS-Stimulated BV2 Cells
3.5. Nec-1/Nec-1s Exerted Antioxidant Effects by Modulating Nrf2/ARE and PKA/CREB Signaling Pathways in LPS-Stimulated BV2 Cells
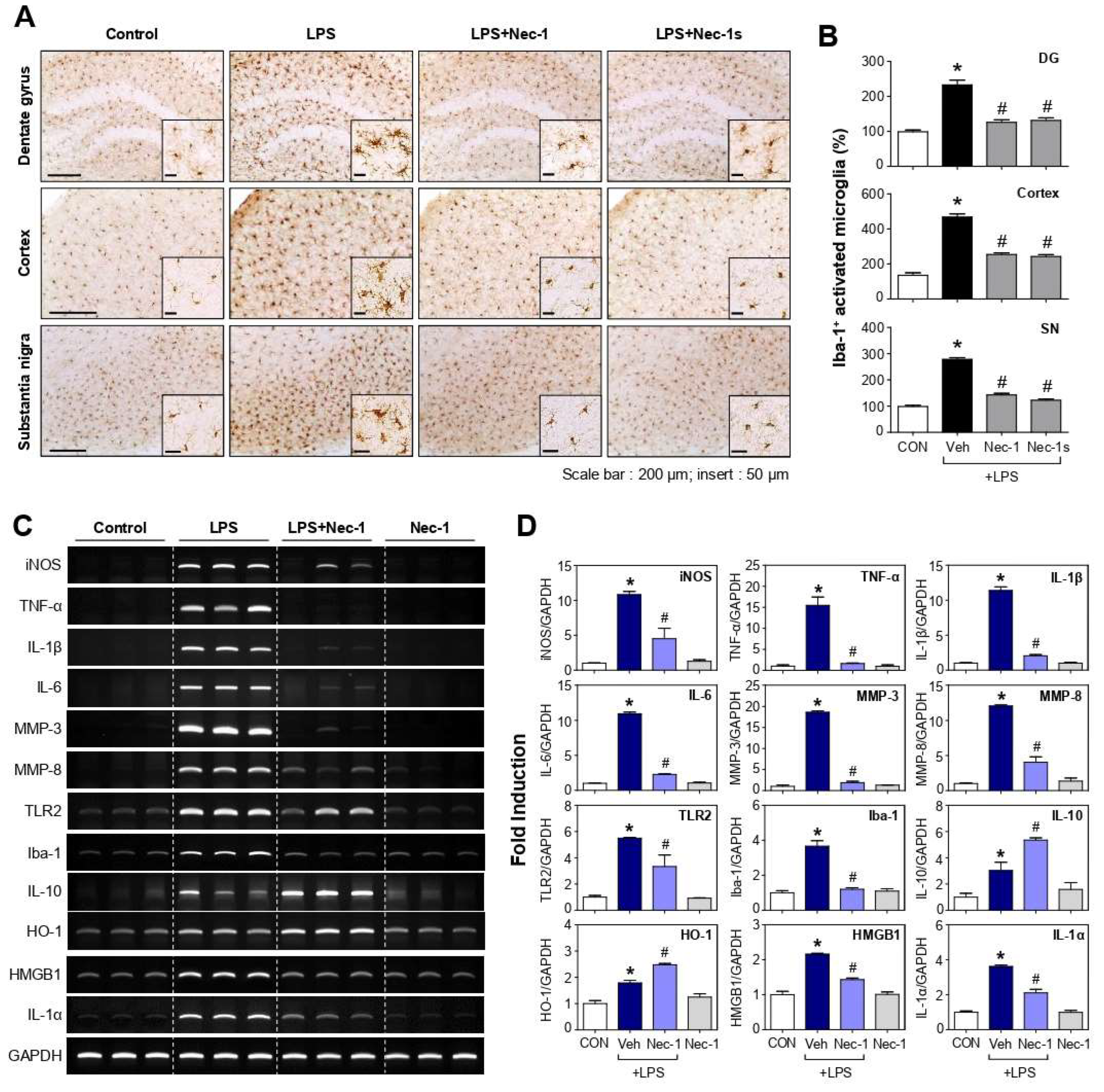
3.6. Nec-1 and Nec-1s Inhibited Microglial Activation and Proinflammatory Gene Expression in the Brains of LPS-Injected Mice
3.7. Nec-1/Nec-1s Inhibited the Phosphorylation and Expression of RIPK1-RIPK3-MLKL in the Brains of LPS-Injected Mice
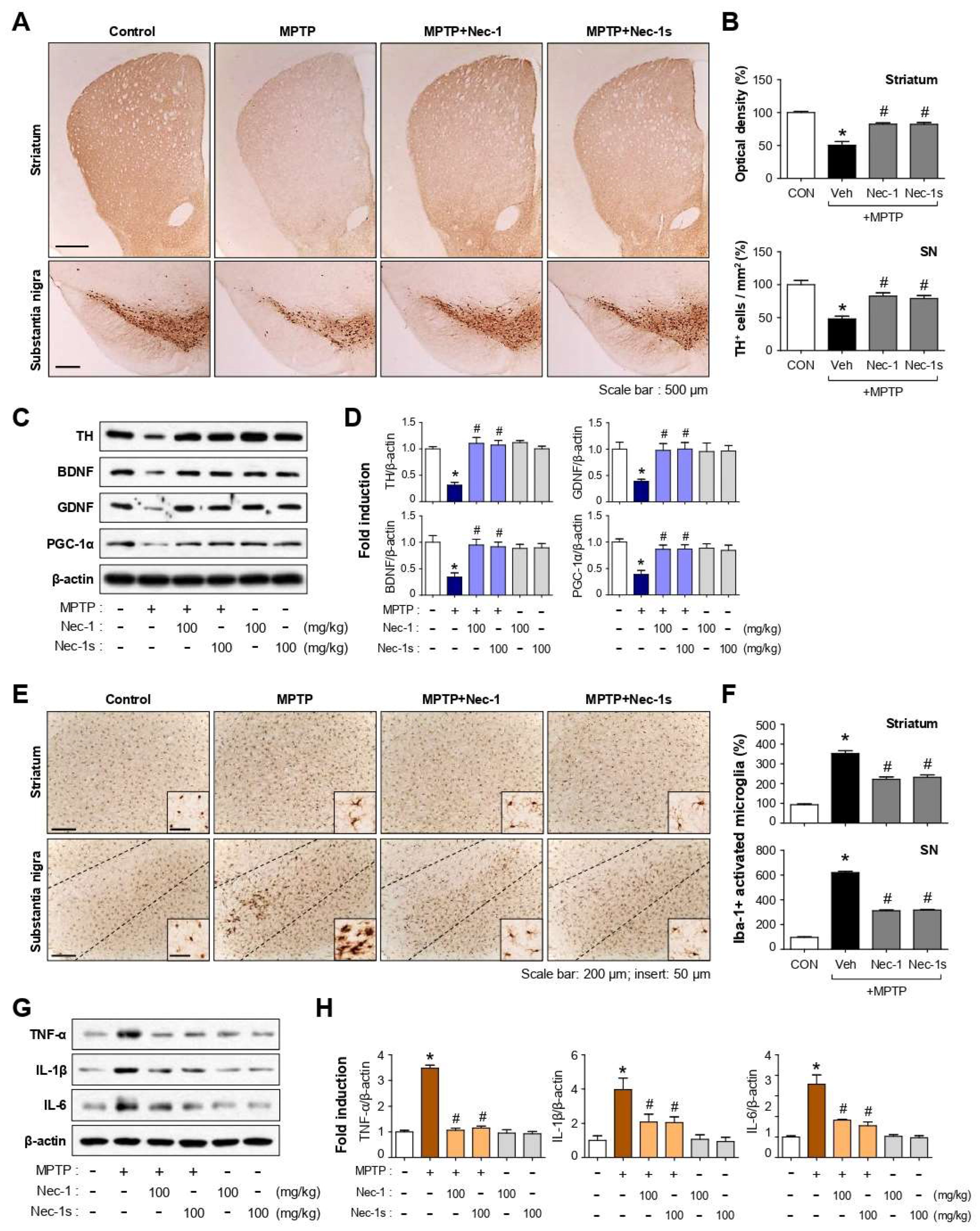
3.8. Nec-1 and Nec-1s Exerted Neuroprotective and Anti-Inflammatory Effects in MPTP-Induced PD Mice
3.9. Nec-1/Nec-1s Reduced p-RIPK1 Expression in Microglia of MPTP-Induced PD Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| ARE | Antioxidant response element |
| cIAP | Cellular inhibitor of apoptosis protein |
| CREB | cAMP response element-binding protein |
| CYLD | Cylindromatosis |
| DAMP | Damage-associated molecular pattern |
| FADD | FAS-associated death domain |
| HMGB1 | High mobility group box 1 |
| HO-1 | Heme oxygenase-1 |
| DO | Indoleamine 2,3-dioxygenase |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MLKL | Mixed lineage kinase domain-like protein |
| MMP | Matrix metalloproteinase |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | Multiple sclerosis |
| Nec-1 | Necrostatin-1 |
| Nec-1s | Necrostatin-1 stable |
| NEMO | NF-κB essential modulator |
| Nrf2 | Nuclear factor-erythroid 2 (NF-E2)-related factor |
| 6-OHDA | 6-hydroxydopamine |
| PD | Parkinson’s disease |
| RIPK | Receptor-interacting protein kinase |
| ROS | Reactive oxygen species |
| SN | Substantia nigra |
| TH | Tyrosine hydroxylase |
| TLR | Toll-like receptor |
| TNFR | Tumor necrosis factor receptor |
References
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Cronk, J.C.; Kipnis, J. Microglia—The brain’s busy bees. F1000Prime Rep. 2013, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, R.J.; Avramopoulos, D.; Jantzie, L.L.; McCallion, A.S. Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases. J. Neuroinflamm. 2022, 19, 223. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson’s disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Molecular mechanisms of necroptosis and relevance for neurodegenerative diseases. Int. Rev. Cell Mol. Biol. 2020, 353, 31–82. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative disesases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festjens, N.; Vanden Berghe, T.; Cornelis, S.; Vandenabeele, P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007, 14, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Degterev, A.; Maki, J.L.; Yuan, J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013, 20, 366. [Google Scholar] [CrossRef] [Green Version]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef] [Green Version]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.R.; Wang, J.; Zhou, S.K.; Yang, L.; Yin, J.L.; Cao, J.P.; Cheng, Y.B. Necrostatin-1 protection of dopaminergic neurons. Neural Regen. Res. 2015, 10, 1120–1124. [Google Scholar]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.S.; Chen, P.; Wang, W.X.; Lin, C.C.; Zhou, Y.; Yu, L.H.; Lin, Y.X.; Xu, Y.F.; Kang, D.Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson’s disease. Lab. Investig. 2020, 100, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Oñate, M.; Catenaccio, A.; Salvadores, N.; Saquel, C.; Martinez, A.; Moreno-Gonzalez, I.; Gamez, N.; Soto, P.; Soto, C.; Hetz, C.; et al. The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 2020, 27, 1169–1185. [Google Scholar] [CrossRef] [Green Version]
- Bocchini, V.; Mazzolla, R.; Barluzzi, R.; Blasi, E.; Sick, P.; Kettenmann, H. An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 1992, 31, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.J.; Lee, E.J.; Park, J.S.; Kim, S.N.; Park, E.M.; Kim, H.S. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: Critical role of PPAR-gamma signaling pathway. Biochem. Pharmacol. 2017, 144, 120–131. [Google Scholar] [CrossRef]
- Lee, E.J.; Ko, H.M.; Jeong, Y.H.; Park, E.M.; Kim, H.S. β-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J. Neuroinflamm. 2015, 12, 133. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.Y.; Park, J.S.; Leem, Y.H.; Park, J.E.; Kim, D.Y.; Choi, Y.H.; Park, E.M.; Kang, J.L.; Kim, H.S. The phosphodiesterase 10 inhibitor papaverine exerts anti-inflammatory and neuroprotective effects via the PKA signaling pathway in neuroinflammation and Parkinson’s disease mouse models. J. Neuroinflamm. 2019, 16, 246. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Park, J.S.; Lee, E.J.; Lee, S.Y.; Kim, D.H.; Kang, J.L.; Kim, H.S. Anti-inflammatory mechanism of ginseng saponin metabolite Rh3 in lipopolysaccharide-stimulated microglia: Critical role of 5’-adenosine monophosphate-activated protein kinase signaling pathway. J. Agric. Food Chem. 2015, 63, 3472–3480. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Park, J.S.; Leem, Y.H.; Kim, D.Y.; Kim, H.S. NQO1 mediates the anti-inflammatory effects of nootkatone in lipopolysaccharide-induced neuroinflammation by modulating the AMPK signaling pathway. Free Radic. Biol. Med. 2021, 164, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wang, F.; Guo, Q.; Li, M.; Wang, L.; Zhang, Z.; Jiang, S.; Jin, H.; Chen, A.; Tan, S.; et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018, 19, 375–387. [Google Scholar] [CrossRef]
- Chen, S.; Lv, X.; Hu, B.; Zhao, L.; Li, S.; Li, Z.; Qing, X.; Liu, H.; Xu, J.; Shao, Z. Critical contribution of RIPK1 mediated mitochondrial dysfunction and oxidative stress to compression-induced rat nucleus pulposus cells necroptosis and apoptosis. Apoptosis 2018, 23, 299–313. [Google Scholar] [CrossRef]
- Laurien, L.; Nagata, M.; Schünke, H.; Delanghe, T.; Wiederstein, J.L.; Kumari, S.; Schwarzer, R.; Corona, T.; Krüger, M.; Bertrand, M.J.M.; et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 2020, 11, 1747. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; DuHadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C.; et al. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012, 3, e437. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A Novel Pathway in Neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 kinases promote cell-death independent inflammation by toll-like receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, S.; Man, H.Y. Bioenergy sensing in the brain: The role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle 2011, 10, 3452–3460. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Zhang, Y.F.; Wang, H.; Ren, R.J.; Cui, H.L.; Huang, W.Y.; Cheng, Q.; Chen, H.Z.; Wang, G. miR-425 deficiency promotes necroptosis and dopaminergic neurodegeneration in Parkinson’s disease. Cell Death Dis. 2019, 10, 589. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| iNOS | CAAGAGTTTGACCAGAGGACC | TGGAACCACTCGTACTTGGGA |
| TNF-α | CCTATGTCTCAGCCTCTTCT | CCTGGTATGAGATAGCAAAT |
| IL-1α | GACCAGCCCGTGTTGC | AGTCCCCGTGCCAGGT |
| IL-1β | GATCCACACTCTCCAGCTGCA | CAACCAACAAGTGATATTCTCCATG |
| IL-6 | CCACTTCACAAGTCGGAGGCTT | CCAGCTTATCTGTTAGGAGA |
| IL-10 | GCCAGTACAGCCGGGAAGACAATA | GCCTTGTAGACACCTTGGTCTT |
| HMGB1 | GCAAAGAAACTAGGAGAGAT | TCTTCTTCATCTTCGTCTTC |
| IL-33 | GAATTCTGCCATGTCTACTG | CCTTGGATGCTCAATGTG |
| MMP-3 | CTCCAGTATTTGTCCTCTAC | TGGAACCACTCGTACTTGGGA |
| MMP-8 | CCAAGGAGTGTCCAAGCCAT | CCTGGTATGAGATAGCAAAT |
| TLR2 | TGCTTTCCTAGCTGGAGATTT | AGTCCCCGTGCCAGGT |
| Iba-1 | AGGAGATTTCAAAAGCTGATGTGG | CAACCAACAAGTGATATTCTCCATG |
| HO-1 | ATACCCGCTACCTGGGTGAC | CCAGCTTATCTGTTAGGAGA |
| p22phox | CAATGGCCAAGCAGACGGTC | GCCTTGTAGACACCTTGGTCTT |
| p47phox | GTTTCAGGTCATCAGGCCGC | TCTTCTTCATCTTCGTCTTC |
| p67phox | AGGCCACTGCAGAGTGCTTG | CCTTGGATGCTCAATGTG |
| gp91phox | TGGCGGTGTGCAGTGCTATC | ATTCAGTCCCTCTATGGA |
| RIPK1 | AGGGTCATGCAGTTTGGAAC | CCTGCAGGAAAACTGCATCG |
| RIPK3 | ATGTCCTGAGAGGCAAGCAC | TGTAACGCAACAGCTTCAGG |
| MLKL | CCCATTTGAAGGCTGTGATT | GTTTGGACGGCAGATCCTCA |
| GAPDH | GGCATGGACTGTGGTCATGA | TGTCACCCTGTGCTTGACCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-Y.; Leem, Y.-H.; Park, J.-S.; Park, J.-E.; Park, J.-M.; Kang, J.L.; Kim, H.-S. RIPK1 Regulates Microglial Activation in Lipopolysaccharide-Induced Neuroinflammation and MPTP-Induced Parkinson’s Disease Mouse Models. Cells 2023, 12, 417. https://doi.org/10.3390/cells12030417
Kim D-Y, Leem Y-H, Park J-S, Park J-E, Park J-M, Kang JL, Kim H-S. RIPK1 Regulates Microglial Activation in Lipopolysaccharide-Induced Neuroinflammation and MPTP-Induced Parkinson’s Disease Mouse Models. Cells. 2023; 12(3):417. https://doi.org/10.3390/cells12030417
Chicago/Turabian StyleKim, Do-Yeon, Yea-Hyun Leem, Jin-Sun Park, Jung-Eun Park, Jae-Min Park, Jihee Lee Kang, and Hee-Sun Kim. 2023. "RIPK1 Regulates Microglial Activation in Lipopolysaccharide-Induced Neuroinflammation and MPTP-Induced Parkinson’s Disease Mouse Models" Cells 12, no. 3: 417. https://doi.org/10.3390/cells12030417