

Th17 Cells, Glucocorticoid Resistance, and Depression

Abstract
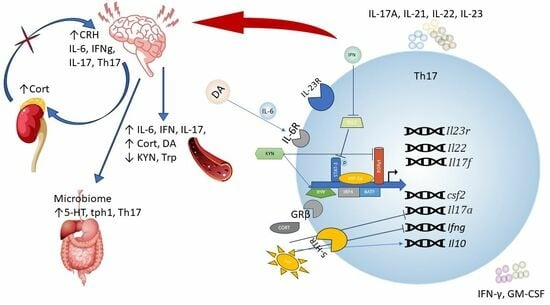
:
1. Introduction
Role of Inflammation and Glucocorticoid Resistance in the Pathogenesis of Depression
2. Th17 Immune Cells and Neuroinflammation
Gut–Brain Axis and Th17 Immune Response
3. For and against Th17 Lymphocyte Involvement in the Pathogenesis of Depression
Effect of Peripheral Biogenic Amines on Th17 Lymphocyte Differentiation
4. Glucocorticoid Resistance and Th17 Lymphocytes
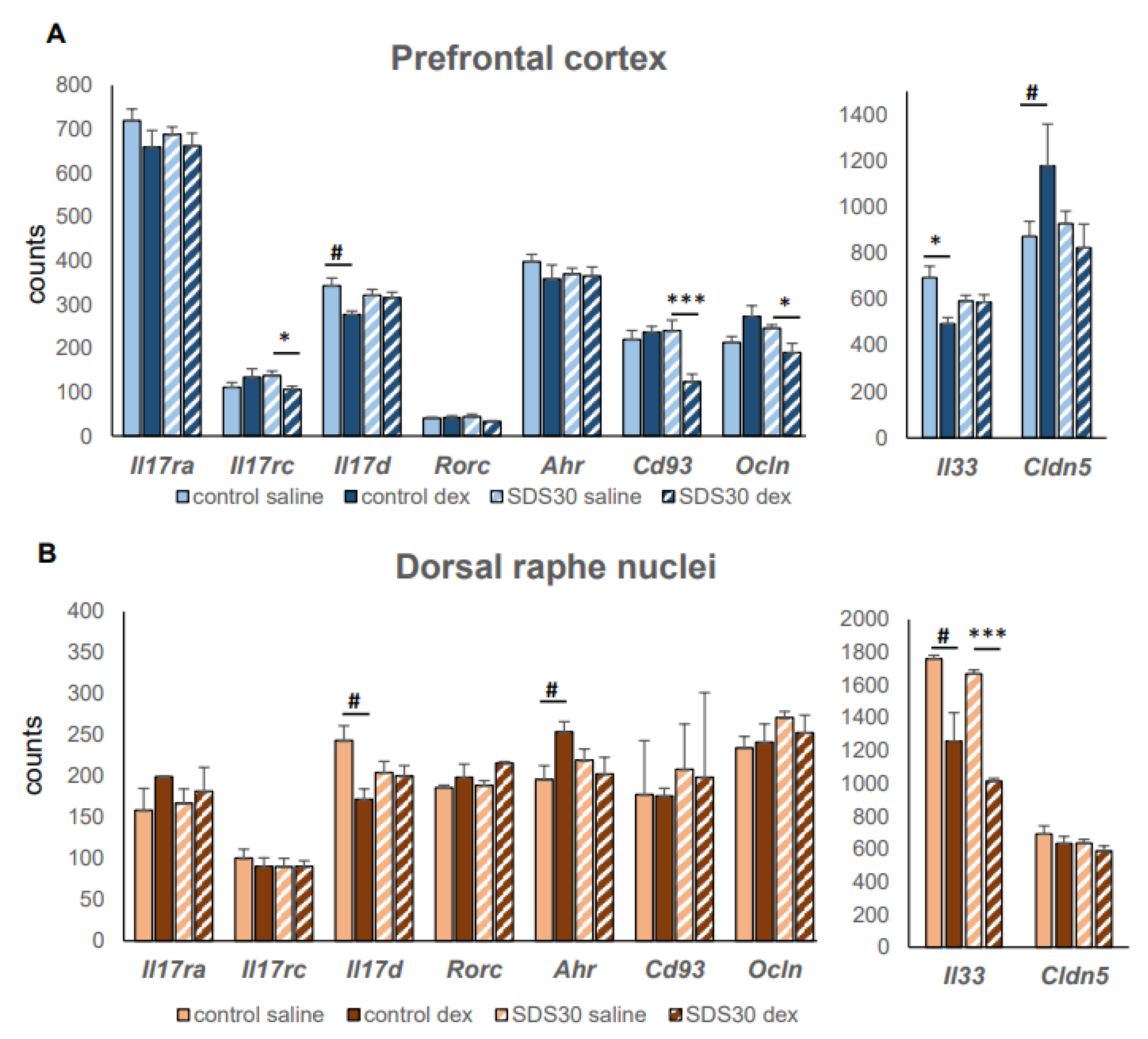
5. Conclusion, Prospects, and Available Statistical Data Linking SDS and Th17
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine, serotonin |
| ACTH | adrenocorticotropin |
| AHR | aryl hydrocarbon receptor |
| BBB | blood–brain barrier |
| CNS | central nervous system |
| CRH | corticotropin-releasing hormone |
| DA | dopamine |
| EAE | experimental autoimmune encephalomyelitis |
| GM-CSF | granulocyte-monocyte colony-stimulating factor |
| GR | glucocorticoid receptor |
| GS | glucocorticoid |
| HPA | hypothalamic-pituitary-adrenal axis |
| IDO | indolamine-2,3-dioxygenase |
| IFN | interferon |
| IL | interleukin |
| MDD | major depression disorders |
| MS | multiple sclerosis |
| PBMC | peripheral blood mononuclear cells |
| RORγt | RAR-related orphan receptor γ |
| TGF-β | transforming growth factor beta |
| Th | T-helper cells |
| TNF | tumor necrosis factor |
| Tph1 | tryptophan hydroxylase 1 |
| Tregs | regulatory T-helper cells |
References
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The Serotonin Theory of Depression: A Systematic Umbrella Review of the Evidence. Mol. Psychiatry 2022, 28, 3243–3256. [Google Scholar] [CrossRef]
- Gałecka, M.; Bliźniewska-Kowalska, K.; Maes, M.; Su, K.-P.; Gałecki, P. Update on the Neurodevelopmental Theory of Depression: Is There Any “Unconscious Code”? Pharmacol. Rep. 2021, 73, 346–356. [Google Scholar] [CrossRef]
- Nothdurfter, C.; Milenkovic, V.M.; Sarubin, N.; Hilbert, S.; Manook, A.; Weigl, J.; Almeqbaali, K.; Wetzel, C.H.; Rupprecht, R.; Baghai, T.C. The Cytokine IL-17A as a Marker of Treatment Resistance in Major Depressive Disorder? Eur. J. Neurosci. 2021, 53, 172–182. [Google Scholar] [CrossRef]
- Banuelos, J.; Cao, Y.; Shin, S.C.; Lu, N.Z. Immunopathology Alters Th17 Cell Glucocorticoid Sensitivity. Allergy 2017, 72, 331–341. [Google Scholar] [CrossRef]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic Regulatory Network Controlling TH17 Cell Differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and Molecular Signature of Pathogenic TH17 Cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- Banuelos, J.; Shin, S.; Cao, Y.; Bochner, B.S.; Morales-Nebreda, L.; Budinger, G.R.S.; Zhou, L.; Li, S.; Xin, J.; Lingen, M.W.; et al. BCL-2 Protects Human and Mouse Th17 Cells from Glucocorticoid-Induced Apoptosis. Allergy 2016, 71, 640–650. [Google Scholar] [CrossRef]
- Schewitz-Bowers, L.P.; Lait, P.J.P.; Copland, D.A.; Chen, P.; Wu, W.; Dhanda, A.D.; Vistica, B.P.; Williams, E.L.; Liu, B.; Jawad, S.; et al. Glucocorticoid-Resistant Th17 Cells Are Selectively Attenuated by Cyclosporine A. Proc. Natl. Acad. Sci. USA 2015, 112, 4080–4085. [Google Scholar] [CrossRef]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; DuPont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 Cells Mediate Steroid-Resistant Airway Inflammation and Airway Hyperresponsiveness in Mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef]
- Carvalho, L.A.; Garner, B.A.; Dew, T.; Fazakerley, H.; Pariante, C.M. Antidepressants, but Not Antipsychotics, Modulate GR Function in Human Whole Blood: An Insight into Molecular Mechanisms. Eur. Neuropsychopharmacol. 2010, 20, 379–387. [Google Scholar] [CrossRef]
- Carvalho, L.A.; Bergink, V.; Sumaski, L.; Wijkhuijs, J.; Hoogendijk, W.J.; Birkenhager, T.K.; Drexhage, H.A. Inflammatory Activation Is Associated with a Reduced Glucocorticoid Receptor Alpha/Beta Expression Ratio in Monocytes of Inpatients with Melancholic Major Depressive Disorder. Transl. Psychiatry 2014, 4, e344. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Suy, E.; Vandervorst, C.; DeJonckheere, C.; Raus, J. Depression-Related Disturbances in Mitogen-Induced Lymphocyte Responses and Interleukin-1? And Soluble Interleukin-2 Receptor Production. Acta Psychiatr. Scand. 1991, 84, 379–386. [Google Scholar] [CrossRef]
- Hearing, S.D.; Norman, M.; Smyth, C.; Foy, C.; Dayan, C.M. Wide Variation in Lymphocyte Steroid Sensitivity Among Healthy Human Volunteers. J. Clin. Endocrinol. Metab. 1999, 84, 4149–4154. [Google Scholar] [CrossRef]
- Bertollo, A.G.; Grolli, R.E.; Plissari, M.E.; Gasparin, V.A.; Quevedo, J.; Réus, G.Z.; Bagatini, M.D.; Ignácio, Z.M. Stress and Serum Cortisol Levels in Major Depressive Disorder: A Cross-Sectional Study. AIMS Neurosci. 2020, 7, 459–469. [Google Scholar] [CrossRef]
- Khantakova, J.N.; Bondar, N.P.; Sapronova, A.A.; Reshetnikov, V.V. Delayed Effects of Neonatal Immune Activation on Brain Neurochemistry and Hypothalamic–Pituitary–Adrenal Axis Functioning. Eur. J. Neurosci. 2022, 56, 5931–5951. [Google Scholar] [CrossRef]
- Perrin, A.J.; Horowitz, M.A.; Roelofs, J.; Zunszain, P.A.; Pariante, C.M. Glucocorticoid Resistance: Is It a Requisite for Increased Cytokine Production in Depression? A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 423. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Merkulova, T.I.; Bondar, N.P. Mechanisms of Brain Glucocorticoid Resistance in Stress-Induced Psychopathologies. Biochemistry 2017, 82, 351–365. [Google Scholar] [CrossRef]
- Black, C.; Miller, B.J. Meta-Analysis of Cytokines and Chemokines in Suicidality: Distinguishing Suicidal versus Nonsuicidal Patients. Biol. Psychiatry 2015, 78, 28–37. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Hiles, S.A.; Baker, A.L.; De Malmanche, T.; Attia, J. Interleukin-6, C-Reactive Protein and Interleukin-10 after Antidepressant Treatment in People with Depression: A Meta-Analysis. Psychol. Med. 2012, 42, 2015–2026. [Google Scholar] [CrossRef]
- Hiles, S.A.; Baker, A.L.; de Malmanche, T.; Attia, J. A Meta-Analysis of Differences in IL-6 and IL-10 between People with and without Depression: Exploring the Causes of Heterogeneity. Brain Behav. Immun. 2012, 26, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Besedovsky, H.O.; Del Rey, A.; Klusman, I.; Furukawa, H.; Monge Arditi, G.; Kabiersch, A. Cytokines as Modulators of the Hypothalamus-Pituitary-Adrenal Axis. J. Steroid. Biochem. Mol. Biol. 1991, 40, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.W.; Hu, F.; Miller, A.H. Cytokine-Effects on Glucocorticoid Receptor Function: Relevance to Glucocorticoid Resistance and the Pathophysiology and Treatment of Major Depression. Brain Behav. Immun. 2007, 21, 9–19. [Google Scholar] [CrossRef]
- Pariante, C.M.; Pearce, B.D.; Pisell, T.L.; Sanchez, C.I.; Po, C.; Su, C.; Miller, A.H. The Proinflammatory Cytokine, Interleukin-1α, Reduces Glucocorticoid Receptor Translocation and Function. Endocrinology 1999, 140, 4359–4366. [Google Scholar] [CrossRef]
- Li, H.; Wang, P.; Zhou, Y.; Zhao, F.; Gao, X.; Wu, C.; Wu, T.; Jiang, L.; Zhang, D. Correlation between Intestinal Microbiotal Imbalance and 5-HT Metabolism, Immune Inflammation in Chronic Unpredictable Mild Stress Male Rats. Genes Brain Behav. 2022, 21, e12806. [Google Scholar] [CrossRef]
- Chang, L.C.; Jamain, S.; Lin, C.W.; Rujescu, D.; Tseng, G.C.; Sibille, E. A Conserved BDNF, Glutamate- and GABA-Enriched Gene Module Related to Human Depression Identified by Coexpression Meta-Analysis and DNA Variant Genome-Wide Association Studies. PLoS ONE 2014, 9, e90980. [Google Scholar] [CrossRef]
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and Brain Excitability. Front. Neuroendocrinol. 2012, 33, 116–125. [Google Scholar] [CrossRef]
- Vezzani, A.; Viviani, B. Neuromodulatory Properties of Inflammatory Cytokines and Their Impact on Neuronal Excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Liu, Q.; Xin, W.; He, P.; Turner, D.; Yin, J.; Gan, Y.; Shi, F.-D.; Wu, J. Interleukin-17 Inhibits Adult Hippocampal Neurogenesis. Sci. Rep. 2014, 4, 7554. [Google Scholar] [CrossRef]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult Hippocampal Neurogenesis Buffers Stress Responses and Depressive Behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef]
- Mahar, I.; Bambico, F.R.; Mechawar, N.; Nobrega, J.N. Stress, Serotonin, and Hippocampal Neurogenesis in Relation to Depression and Antidepressant Effects. Neurosci. Biobehav. Rev. 2014, 38, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 Rather than Interleukin-12 Is the Critical Cytokine for Autoimmune Inflammation of the Brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 Drives a Pathogenic T Cell Population That Induces Autoimmune Inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Kuchroo, V.K. IL-12– and IL-23–Induced T Helper Cell Subsets. J. Exp. Med. 2005, 201, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Cerboni, S.; Gehrmann, U.; Preite, S.; Mitra, S. Cytokine-regulated Th17 Plasticity in Human Health and Diseases. Immunology 2021, 163, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Dong, C. TH17 Cells in Development: An Updated View of Their Molecular Identity and Genetic Programming. Nat. Rev. Immunol. 2008, 8, 337–348. [Google Scholar] [CrossRef]
- Bedoya, S.K.; Wilson, T.D.; Collins, E.L.; Lau, K.; Larkin, J. Isolation and Th17 Differentiation of Naïve CD4 T Lymphocytes. J. Vis. Exp. 2013, 79, e50765. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-β and IL-6 Drive the Production of IL-17 and IL-10 by T Cells and Restrain TH-17 Cell–Mediated Pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef]
- Schnell, A.; Littman, D.R.; Kuchroo, V.K. TH17 Cell Heterogeneity and Its Role in Tissue Inflammation. Nat. Immunol. 2023, 24, 19–29. [Google Scholar] [CrossRef]
- Moser, T.; Akgün, K.; Proschmann, U.; Sellner, J.; Ziemssen, T. The Role of TH17 Cells in Multiple Sclerosis: Therapeutic Implications. Autoimmun. Rev. 2020, 19, 102647. [Google Scholar] [CrossRef] [PubMed]
- Schnell, A.; Huang, L.; Singer, M.; Singaraju, A.; Barilla, R.M.; Regan, B.M.L.; Bollhagen, A.; Thakore, P.I.; Dionne, D.; Delorey, T.M.; et al. Stem-like Intestinal Th17 Cells Give Rise to Pathogenic Effector T Cells during Autoimmunity. Cell 2021, 184, 6281–6298.e23. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Perera, D.N.; Fan, H.; Russ, B.E.; Harris, J.; Morand, E.F. GILZ Regulates Th17 Responses and Restrains IL-17-Mediated Skin Inflammation. J. Autoimmun. 2015, 61, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Bussi, C.; Metidji, A.; Iseppon, A.; Lee, S.; Tolaini, M.; Li, Y.; Kelly, G.; Chakravarty, P.; Shoaie, S.; et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 2019, 51, 77–89.e6. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Omenetti, S. The Dichotomous Nature of T Helper 17 Cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a Cross-Talk between Human Neutrophils and Th17 Cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.-P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of Pathogenic TH17 Cells in the Absence of TGF-β Signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-Induced Human TH17 Cells Produce IFN-γ or IL-10 and Are Regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef]
- Thakore, P.I.; Schnell, A.; Zhao, M.; Huang, L.; Hou, Y.; Christian, E.; Zaghouani, S.; Wang, C.; Singh, V.; Ma, S.; et al. The Chromatin Landscape of Th17 Cells Reveals Mechanisms of Diversification of Regulatory and Pro-Inflammatory States. BioRxiv 2022, 02.26.482041. [Google Scholar] [CrossRef]
- Khantakova, J.; Sennikov, S. T-Helper Cells Flexibility: The Possibility of Reprogramming T Cells Fate. Front. Immunol. T Cell Biol. 2023, 14, 1284178. [Google Scholar] [CrossRef]
- Kebir, H.; Ifergan, I.; Alvarez, J.I.; Bernard, M.; Poirier, J.; Arbour, N.; Duquette, P.; Prat, A. Preferential Recruitment of Interferon-γ-Expressing T H 17 Cells in Multiple Sclerosis. Ann. Neurol. 2009, 66, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C.; Tolaini, M.; Menzel, U.; et al. Fate Mapping of IL-17-Producing T Cells in Inflammatory Responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef] [PubMed]
- McGinley, A.M.; Sutton, C.E.; Edwards, S.C.; Leane, C.M.; DeCourcey, J.; Teijeiro, A.; Hamilton, J.A.; Boon, L.; Djouder, N.; Mills, K.H.G. Interleukin-17A Serves a Priming Role in Autoimmunity by Recruiting IL-1β-Producing Myeloid Cells That Promote Pathogenic T Cells. Immunity 2020, 52, 342–356.e6. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.E.; Papadopoulos, A.; Poon, L.; Perks, P.; Lightman, S.L.; Checkley, S.; Shanks, N. Dexamethasone-Induced Effects on Lymphocyte Distribution and Expression of Adhesion Molecules in Treatment-Resistant Depression. Psychiatry Res. 2002, 113, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, A. Neutrophils: Cinderella of Innate Immune System. Int. Immunopharmacol. 2010, 10, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Udina, M.; Castellví, P.; Moreno-España, J.; Navinés, R.; Valdés, M.; Forns, X.; Langohr, K.; Solà, R.; Vieta, E.; Martín-Santos, R. Interferon-Induced Depression in Chronic Hepatitis C. J. Clin. Psychiatry 2012, 73, 1128–1138. [Google Scholar] [CrossRef]
- Mostafavi, S.; Battle, A.; Zhu, X.; Potash, J.B.; Weissman, M.M.; Shi, J.; Beckman, K.; Haudenschild, C.; McCormick, C.; Mei, R.; et al. Type I Interferon Signaling Genes in Recurrent Major Depression: Increased Expression Detected by Whole-Blood RNA Sequencing. Mol. Psychiatry 2014, 19, 1267–1274. [Google Scholar] [CrossRef]
- Tamassia, N.; Arruda-Silva, F.; Wright, H.L.; Moots, R.J.; Gardiman, E.; Bianchetto-Aguilera, F.; Gasperini, S.; Capone, M.; Maggi, L.; Annunziato, F.; et al. Human Neutrophils Activated via TLR8 Promote Th17 Polarization through IL-23. J. Leukoc. Biol. 2019, 105, 1155–1165. [Google Scholar] [CrossRef]
- Mourik, B.C.; Lubberts, E.; de Steenwinkel, J.E.M.; Ottenhoff, T.H.M.; Leenen, P.J.M. Interactions between Type 1 Interferons and the Th17 Response in Tuberculosis: Lessons Learned from Autoimmune Diseases. Front. Immunol. 2017, 8, 294. [Google Scholar] [CrossRef]
- Gaffen, S.L. Structure and Signalling in the IL-17 Receptor Family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef]
- Weaver, C.T.; Hatton, R.D.; Mangan, P.R.; Harrington, L.E. IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages. Annu. Rev. Immunol. 2007, 25, 821–852. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional Specialization of Interleukin-17 Family Members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Zaph, C.; Du, Y.; Saenz, S.A.; Nair, M.G.; Perrigoue, J.G.; Taylor, B.C.; Troy, A.E.; Kobuley, D.E.; Kastelein, R.A.; Cua, D.J.; et al. Commensal-Dependent Expression of IL-25 Regulates the IL-23–IL-17 Axis in the Intestine. J. Exp. Med. 2008, 205, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bechara, R.; Zhao, J.; McGeachy, M.J.; Gaffen, S.L. IL-17 Receptor–Based Signaling and Implications for Disease. Nat. Immunol. 2019, 20, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, A.; Lehmann, S.; Blank, J.; Kolbinger, F.; Rondeau, J.-M. Structural Analysis Reveals That the Cytokine IL-17F Forms a Homodimeric Complex with Receptor IL-17RC to Drive IL-17RA-Independent Signaling. Immunity 2020, 52, 499–512.e5. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 Receptor D Constitutes an Alternative Receptor for Interleukin-17A Important in Psoriasis-like Skin Inflammation. Sci. Immunol. 2019, 4, eaau9657. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Hauptmann, J.; Regen, T. The Role of IL-17 in CNS Diseases. Acta Neuropathol 2015, 129, 625–637. [Google Scholar] [CrossRef]
- Regen, T.; Isaac, S.; Amorim, A.; Núñez, N.G.; Hauptmann, J.; Shanmugavadivu, A.; Klein, M.; Sankowski, R.; Mufazalov, I.A.; Yogev, N.; et al. IL-17 Controls Central Nervous System Autoimmunity through the Intestinal Microbiome. Sci. Immunol. 2021, 6, eaaz6563. [Google Scholar] [CrossRef]
- Peng, Z.; Peng, S.; Lin, K.; Zhao, B.; Wei, L.; Tuo, Q.; Liao, D.; Yuan, T.; Shi, Z. Chronic Stress-Induced Depression Requires the Recruitment of Peripheral Th17 Cells into the Brain. J. Neuroinflamm. 2022, 19, 186. [Google Scholar] [CrossRef]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular Mechanisms of IL-17-induced Blood-brain Barrier Disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 Lymphocytes Promote Blood-Brain Barrier Disruption and Central Nervous System Inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- van Langelaar, J.; van der Vuurst de Vries, R.M.; Janssen, M.; Wierenga-Wolf, A.F.; Spilt, I.M.; Siepman, T.A.; Dankers, W.; Verjans, G.M.G.M.; de Vries, H.E.; Lubberts, E.; et al. T Helper 17.1 Cells Associate with Multiple Sclerosis Disease Activity: Perspectives for Early Intervention. Brain 2018, 141, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Dong, H.; Wang, Y.; Zhou, Q.; Xu, M.; Qian, Y.; Sun, J. IL-17A Contributes to Perioperative Neurocognitive Disorders through Blood-Brain Barrier Disruption in Aged Mice. J. Neuroinflamm. 2018, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- Kawanokuchi, J.; Shimizu, K.; Nitta, A.; Yamada, K.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Production and Functions of IL-17 in Microglia. J. Neuroimmunol. 2008, 194, 54–61. [Google Scholar] [CrossRef]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Colic, M. IL-17 Signalling in Astrocytes Promotes Glutamate Excitotoxicity: Indications for the Link between Inflammatory and Neurodegenerative Events in Multiple Sclerosis. Mult. Scler. Relat. Disord 2017, 11, 12–17. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-Producing Cells in Protection versus Pathology. Nat. Rev. Immunol. 2023, 23, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Kubick, N.; Klimovich, P.; Flournoy, P.H.; Bieńkowska, I.; Łazarczyk, M.; Sacharczuk, M.; Bhaumik, S.; Mickael, M.-E.; Basu, R. Interleukins and Interleukin Receptors Evolutionary History and Origin in Relation to CD4+ T Cell Evolution. Genes 2021, 12, 813. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface Phenotype and Antigenic Specificity of Human Interleukin 17–Producing T Helper Memory Cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef]
- Sallusto, F. Heterogeneity of Human CD4+ T Cells Against Microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Westfall, S.; Caracci, F.; Zhao, D.; Wu, Q.; Frolinger, T.; Simon, J.; Pasinetti, G.M. Microbiota Metabolites Modulate the T Helper 17 to Regulatory T Cell (Th17/Treg) Imbalance Promoting Resilience to Stress-Induced Anxiety- and Depressive-like Behaviors. Brain Behav. Immun. 2021, 91, 350–368. [Google Scholar] [CrossRef]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a Social Stressor Alters the Structure of the Intestinal Microbiota: Implications for Stressor-Induced Immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Westfall, S.; Caracci, F.; Estill, M.; Frolinger, T.; Shen, L.; Pasinetti, G.M. Chronic Stress-Induced Depression and Anxiety Priming Modulated by Gut-Brain-Axis Immunity. Front. Immunol. 2021, 12, 2474. [Google Scholar] [CrossRef] [PubMed]
- Barandouzi, Z.A.; Starkweather, A.R.; Henderson, W.A.; Gyamfi, A.; Cong, X.S. Altered Composition of Gut Microbiota in Depression: A Systematic Review. Front. Psychiatry 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.A.; Diaz-Arteche, C.; Eliby, D.; Schwartz, O.S.; Simmons, J.G.; Cowan, C.S.M. The Gut Microbiota in Anxiety and Depression—A Systematic Review. Clin. Psychol. Rev. 2021, 83, 101943. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered Fecal Microbiota Composition in Patients with Major Depressive Disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, M.; Wang, Y.; Dorfman, R.G.; Liu, H.; Yu, T.; Chen, X.; Tang, D.; Xu, L.; Yin, Y.; et al. Faecalibacterium Prausnitzii Produces Butyrate to Maintain Th17/Treg Balance and to Ameliorate Colorectal Colitis by Inhibiting Histone Deacetylase 1. Inflamm. Bowel Dis. 2018, 24, 1926–1940. [Google Scholar] [CrossRef]
- Huang, F.; Qiao, H.; Yin, J.; Gao, Y.; Ju, Y.; Li, Y. Early-Life Exposure to Clostridium Leptum Causes Pulmonary Immunosuppression. PLoS ONE 2015, 10, e0141717. [Google Scholar] [CrossRef]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-Cell Responses to Gut Microbiota Promote Experimental Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 (Supp. 1), 4615–4622. [Google Scholar] [CrossRef]
- Miyauchi, E.; Kim, S.-W.; Suda, W.; Kawasumi, M.; Onawa, S.; Taguchi-Atarashi, N.; Morita, H.; Taylor, T.D.; Hattori, M.; Ohno, H. Gut Microorganisms Act Together to Exacerbate Inflammation in Spinal Cords. Nature 2020, 585, 102–106. [Google Scholar] [CrossRef]
- Gödel, C.; Kunkel, B.; Kashani, A.; Lassmann, H.; Arumugam, M.; Krishnamoorthy, G. Perturbation of Gut Microbiota Decreases Susceptibility but Does Not Modulate Ongoing Autoimmune Neurological Disease. J. Neuroinflamm. 2020, 17, 79. [Google Scholar] [CrossRef]
- Alvarez-Mon, M.A.; Gómez-Lahoz, A.M.; Orozco, A.; Lahera, G.; Diaz, D.; Ortega, M.A.; Albillos, A.; Quintero, J.; Aubá, E.; Monserrat, J.; et al. Expansion of CD4 T Lymphocytes Expressing Interleukin 17 and Tumor Necrosis Factor in Patients with Major Depressive Disorder. J. Pers. Med. 2021, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Villanueva, E.; Pérez-Sánchez, G.; Alvarez-Herrera, S.; Girón-Pérez, M.I.; Arreola, R.; Cruz-Fuentes, C.; Palacios, L.; de la Peña, F.R.; Pavón, L. Alterations in the Levels of Growth Factors in Adolescents with Major Depressive Disorder: A Longitudinal Study during the Treatment with Fluoxetine. Mediat. Inflamm. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, T.; Chen, P.; Ouyang, J.; Xu, G.; Zeng, Z.; Sun, Y. Emerging Tendency towards Autoimmune Process in Major Depressive Patients: A Novel Insight from Th17 Cells. Psychiatry Res. 2011, 188, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Davami, M.; Baharlou, R.; Ahmadi Vasmehjani, A.; Ghanizadeh, A.; Keshtkar, M.; Dezhkam, I.; Atashzar, M. Elevated IL-17 and TGF-β Serum Levels: A Positive Correlation between T-Helper 17 Cell-Related Pro-Inflammatory Responses with Major Depressive Disorder. Basic Clin. Neurosci. J. 2016, 7, 137–142. [Google Scholar] [CrossRef]
- do Sacramento, P.M.; Sales, M.; Kasahara, T.d.M.; Monteiro, C.; Oyamada, H.; Dias, A.S.O.; Lopes, L.; Castro, C.T.; Rossi, Á.D.; Milioni, L.M.; et al. Major Depression Favors the Expansion of Th17-like Cells and Decrease the Proportion of CD39+Treg Cell Subsets in Response to Myelin Antigen in Multiple Sclerosis Patients. Cell. Mol. Life Sci. 2022, 79, 298. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Kumar, P.K.; Mitra, P.; Purohit, P.; Nebhinani, N.; Sharma, P. Circulating T Helper 17 and IFN-γ Positive Th17 Cells in Major Depressive Disorder. Behav. Brain Res. 2020, 394, 112811. [Google Scholar] [CrossRef]
- Ghosh, R.; Mitra, P.; Kumar, P.V.S.N.K.; Goyal, T.; Sharma, P. T Helper Cells in Depression: Central Role of Th17 Cells. Crit. Rev. Clin. Lab. Sci. 2022, 59, 19–39. [Google Scholar] [CrossRef]
- Jha, M.K.; Miller, A.H.; Minhajuddin, A.; Trivedi, M.H. Association of T and Non-T Cell Cytokines with Anhedonia: Role of Gender Differences. Psychoneuroendocrinology 2018, 95, 1–7. [Google Scholar] [CrossRef]
- Jha, M.K.; Minhajuddin, A.; Gadad, B.S.; Greer, T.L.; Mayes, T.L.; Trivedi, M.H. Interleukin 17 Selectively Predicts Better Outcomes with Bupropion-SSRI Combination: Novel T Cell Biomarker for Antidepressant Medication Selection. Brain Behav. Immun. 2017, 66, 103–110. [Google Scholar] [CrossRef]
- Leff Gelman, P.; Mancilla-Herrera, I.; Flores-Ramos, M.; Saravia Takashima, M.F.; Cruz Coronel, F.M.; Cruz Fuentes, C.; Pérez Molina, A.; Hernández-Ruiz, J.; Silva-Aguilera, F.S.; Farfan-Labonne, B.; et al. The Cytokine Profile of Women with Severe Anxiety and Depression during Pregnancy. BMC Psychiatry 2019, 19, 104. [Google Scholar] [CrossRef]
- Min, Z.; Li, Y.; Ying, H. Blood T-helper 17 Cells and Interleukin-17A Correlate with the Elevated Risk of Postpartum Depression and Anxiety. J. Clin. Lab. Anal. 2022, 36, e24559. [Google Scholar] [CrossRef]
- Oyamada, H.A.A.; Cafasso, M.O.S.D.; Vollmer, C.M.; Alvim, F.; Lopes, L.M.; Castro, C.; Sacramento, P.M.; Sales, M.C.; Kasahara, T.M.; Linhares, U.C.; et al. Major Depressive Disorder Enhances Th2 and Th17 Cytokines in Patients Suffering from Allergic Rhinitis and Asthma. Int. Arch. Allergy Immunol. 2021, 182, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Primo de Carvalho Alves, L.; Sica da Rocha, N. Different Cytokine Patterns Associate with Melancholia Severity among Inpatients with Major Depressive Disorder. Ther. Adv. Psychopharmacol. 2020, 10, 204512532093792. [Google Scholar] [CrossRef] [PubMed]
- Sacramento, P.M.; Monteiro, C.; Dias, A.S.O.; Kasahara, T.M.; Ferreira, T.B.; Hygino, J.; Wing, A.C.; Andrade, R.M.; Rueda, F.; Sales, M.C.; et al. Serotonin Decreases the Production of Th1/Th17 Cytokines and Elevates the Frequency of Regulatory CD4+ T-Cell Subsets in Multiple Sclerosis Patients. Eur. J. Immunol. 2018, 48, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Schiweck, C.; Valles-Colomer, M.; Arolt, V.; Müller, N.; Raes, J.; Wijkhuijs, A.; Claes, S.; Drexhage, H.; Vrieze, E. Depression and Suicidality: A Link to Premature T Helper Cell Aging and Increased Th17 Cells. Brain Behav. Immun. 2020, 87, 603–609. [Google Scholar] [CrossRef]
- Şimşek, Ş.; Yüksel, T.; Çim, A.; Kaya, S. Serum Cytokine Profiles of Children with Obsessive-Compulsive Disorder Shows the Evidence of Autoimmunity. Int. J. Neuropsychopharmacol. 2016, 19, pyw027. [Google Scholar] [CrossRef]
- Hennings, J.M.; Uhr, M.; Klengel, T.; Weber, P.; Pütz, B.; Touma, C.; Czamara, D.; Ising, M.; Holsboer, F.; Lucae, S. RNA Expression Profiling in Depressed Patients Suggests Retinoid-Related Orphan Receptor Alpha as a Biomarker for Antidepressant Response. Transl. Psychiatry 2015, 5, e538. [Google Scholar] [CrossRef]
- Rodríguez, N.; Morer, A.; González-Navarro, E.A.; Serra-Pages, C.; Boloc, D.; Torres, T.; Martinez-Pinteño, A.; Mas, S.; Lafuente, A.; Gassó, P.; et al. Altered Frequencies of Th17 and Treg Cells in Children and Adolescents with Obsessive-Compulsive Disorder. Brain Behav. Immun. 2019, 81, 608–616. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, Z.; Ni, L.; Cao, J.; Tan, M.; Wu, X.; Xu, Y.; Hu, J. Correlation between Anxiety-Depression Symptoms and Immune Characteristics in Inpatients with 2019 Novel Coronavirus in Wuhan, China. J. Psychiatr. Res. 2021, 141, 378–384. [Google Scholar] [CrossRef]
- Rossi, G.N.; Hallak, J.E.C.; Baker, G.; Dursun, S.M.; dos Santos, R.G. The Effects of Ketamine and Classic Hallucinogens on Neurotrophic and Inflammatory Markers in Unipolar Treatment-Resistant Depression: A Systematic Review of Clinical Trials. Eur. Arch. Psychiatry Clin. Neurosci. 2023, 273, 129–155. [Google Scholar] [CrossRef]
- Cui, M.; Dai, W.; Kong, J.; Chen, H. Th17 Cells in Depression: Are They Crucial for the Antidepressant Effect of Ketamine? Front. Pharmacol. 2021, 12, 649144. [Google Scholar] [CrossRef] [PubMed]
- Saraykar, S.; Cao, B.; Barroso, L.S.; Pereira, K.S.; Bertola, L.; Nicolau, M.; Ferreira, J.D.; Dias, N.S.; Vieira, E.L.; Teixeira, A.L.; et al. Plasma IL-17A Levels in Patients with Late-Life Depression. Rev. Bras. De Psiquiatr. 2017, 40, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Kim, Y.-K.; Hwang, J.-A.; Yoon, H.-K.; Ko, Y.-H.; Han, C.; Lee, H.-J.; Ham, B.-J.; Lee, H.S. Plasma Levels of IL-23 and IL-17 before and after Antidepressant Treatment in Patients with Major Depressive Disorder. Psychiatry Investig 2013, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Fam, J.; Rush, A.J.; Burt, T.; Chan, E.S.; Siddiqui, F.J.; Assam, P.N.; Lai, O.F.; Chan, H.N.; Ng, B.Y.; Khoo, D.H. Thyroid Autoimmune Antibodies and Major Depressive Disorder in Women. Ann. Acad. Med. Singap. 2015, 44, 284–289. [Google Scholar] [CrossRef]
- Kim, J.; Suh, Y.H.; Chang, K.A. Interleukin-17 Induced by Cumulative Mild Stress Promoted Depression-like Behaviors in Young Adult Mice. Mol. Brain 2021, 14, 1–13. [Google Scholar] [CrossRef]
- Beurel, E.; Harrington, L.E.; Jope, R.S. Inflammatory T Helper 17 Cells Promote Depression-like Behavior in Mice. Biol. Psychiatry 2013, 73, 622–630. [Google Scholar] [CrossRef]
- Kim, S.-J.; Lee, H.; Lee, G.; Oh, S.-J.; Shin, M.-K.; Shim, I.; Bae, H. CD4+CD25+ Regulatory T Cell Depletion Modulates Anxiety and Depression-Like Behaviors in Mice. PLoS ONE 2012, 7, e42054. [Google Scholar] [CrossRef]
- Khantakova, J.N.; Bondar, N.P.; Antontseva, E.V.; Reshetnikov, V.V. Once Induced, It Lasts for a Long Time: The Structural and Molecular Signatures Associated with Depressive-like Behavior after Neonatal Immune Activation. Front. Cell Neurosci. 2022, 16, 1066794. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Fardan, A.S.; El-Sherbeeny, A.M.; Ibrahim, K.E.; Attia, S.M. IL-17A Causes Depression-like Symptoms via NFκB and P38MAPK Signaling Pathways in Mice: Implications for Psoriasis Associated Depression. Cytokine 2017, 97, 14–24. [Google Scholar] [CrossRef]
- Alves de Lima, K.; Rustenhoven, J.; Da Mesquita, S.; Wall, M.; Salvador, A.F.; Smirnov, I.; Martelossi Cebinelli, G.; Mamuladze, T.; Baker, W.; Papadopoulos, Z.; et al. Meningeal Γδ T Cells Regulate Anxiety-like Behavior via IL-17a Signaling in Neurons. Nat. Immunol. 2020, 21, 1421–1429. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, S.; Lu, Y.; Wang, Y.; Zhao, J.; Li, L. T Cell Responses in Depressed Mice Induced by Chronic Unpredictable Mild Stress. J. Affect. Disord. 2022, 296, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Bondar, N.; Bryzgalov, L.; Ershov, N.; Gusev, F.; Reshetnikov, V.; Avgustinovich, D.; Tenditnik, M.; Rogaev, E.; Merkulova, T. Molecular Adaptations to Social Defeat Stress and Induced Depression in Mice. Mol. Neurobiol. 2018, 55, 3394–3407. [Google Scholar] [CrossRef] [PubMed]
- Reshetnikov, V.V.; Kisaretova, P.E.; Bondar, N.P. Transcriptome Alterations Caused by Social Defeat Stress of Various Durations in Mice and Its Relevance to Depression and Posttraumatic Stress Disorder in Humans: A Meta-Analysis. Int. J. Mol. Sci. 2022, 23, 13792. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lee, H.; Zhao, X.; Han, J.; Su, Y.; Sun, Q.; Shao, J.; Ge, J.; Zhao, Y.; Bai, X.; et al. Interleukin-17D Regulates Group 3 Innate Lymphoid Cell Function through Its Receptor CD93. Immunity 2021, 54, 673–686.e4. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Luebbe, J.; Kilian, C.; Riedel, J.-H.; Hiekmann, S.; Asada, N.; Ginsberg, P.; Robben, L.; Song, N.; Kaffke, A.; et al. IL-17 Receptor C Signaling Controls CD4+ TH17 Immune Responses and Tissue Injury in Immune-Mediated Kidney Diseases. J. Am. Soc. Nephrol. 2021, 32, 3081–3098. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ota, N.; Peng, I.; Refino, C.J.; Danilenko, D.M.; Caplazi, P.; Ouyang, W. IL-17RC Is Required for IL-17A– and IL-17F–Dependent Signaling and the Pathogenesis of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2010, 184, 4307–4316. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, C.; Zeng, X.; Resch, J.M.; Jedrychowski, M.P.; Yang, Z.; Desai, B.N.; Banks, A.S.; Lowell, B.B.; Mathis, D.; et al. Γδ T Cells and Adipocyte IL-17RC Control Fat Innervation and Thermogenesis. Nature 2020, 578, 610–614. [Google Scholar] [CrossRef]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-1 Leads to Increased Neuronal Cell Death and Microgliosis in Mouse Brain Initiate or Resolve Inflammation (Dennler et al Quently, Disruption of TGF-s Results in Severe Isoform. Roberts Sporn 2003, 40, 1133–1145. [Google Scholar] [CrossRef]
- Yan, C.; Lei, Y.; Lin, T.-J.; Hoskin, D.W.; Ma, A.; Wang, J. IL-17RC Is Critically Required to Maintain Baseline A20 Production to Repress JNK Isoform-Dependent Tumor-Specific Proliferation. Oncotarget 2017, 8, 43153–43168. [Google Scholar] [CrossRef]
- Pascual-Reguant, A.; Bayat Sarmadi, J.; Baumann, C.; Noster, R.; Cirera-Salinas, D.; Curato, C.; Pelczar, P.; Huber, S.; Zielinski, C.E.; Löhning, M.; et al. TH17 Cells Express ST2 and Are Controlled by the Alarmin IL-33 in the Small Intestine. Mucosal Immunol. 2017, 10, 1431–1442. [Google Scholar] [CrossRef]
- Welcome, M.O. Cellular Mechanisms and Molecular Signaling Pathways in Stress-Induced Anxiety, Depression, and Blood–Brain Barrier Inflammation and Leakage. Inflammopharmacology 2020, 28, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Holck, A.; Wolkowitz, O.M.; Mellon, S.H.; Reus, V.I.; Nelson, J.C.; Westrin, Å.; Lindqvist, D. Plasma Serotonin Levels Are Associated with Antidepressant Response to SSRIs. J. Affect. Disord. 2019, 250, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Drevets, W.; Turecki, G.; Li, Q.S. The Relationship between Plasma Serotonin and Kynurenine Pathway Metabolite Levels and the Treatment Response to Escitalopram and Desvenlafaxine. Brain Behav. Immun. 2020, 87, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Dvojkovic, A.; Nikolac Perkovic, M.; Sagud, M.; Nedic Erjavec, G.; Mihaljevic Peles, A.; Svob Strac, D.; Vuksan Cusa, B.; Tudor, L.; Kusevic, Z.; Konjevod, M.; et al. Effect of Vortioxetine vs. Escitalopram on Plasma BDNF and Platelet Serotonin in Depressed Patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 105, 110016. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Herr, D.; MacIver, N.J.; Rathmell, J.C.; Gerriets, V.A. CD4 T Cells Differentially Express Cellular Machinery for Serotonin Signaling, Synthesis, and Metabolism. Int. Immunopharmacol. 2020, 88, 106922. [Google Scholar] [CrossRef]
- Sviridova, A.; Rogovskii, V.; Kudrin, V.; Pashenkov, M.; Boyko, A.; Melnikov, M. The Role of 5-HT2B-Receptors in Fluoxetine-Mediated Modulation of Th17- and Th1-Cells in Multiple Sclerosis. J. Neuroimmunol. 2021, 356, 577608. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, B.; Qiu, W.; Yang, L.; Hu, B.; Tian, X.; Yang, H. Altered Expression of CD4+CD25+ Regulatory T Cells and Its 5-HT1a Receptor in Patients with Major Depression Disorder. J. Affect. Disord. 2010, 124, 68–75. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The Role of Inflammation in Depression: From Evolutionary Imperative to Modern Treatment Target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Arnone, D.; Saraykar, S.; Salem, H.; Teixeira, A.L.; Dantzer, R.; Selvaraj, S. Role of Kynurenine Pathway and Its Metabolites in Mood Disorders: A Systematic Review and Meta-Analysis of Clinical Studies. Neurosci. Biobehav. Rev. 2018, 92, 477–485. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the Mammalian Brain: When Physiology Meets Pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Almulla, A.F.; Thipakorn, Y.; Vasupanrajit, A.; Abo Algon, A.A.; Tunvirachaisakul, C.; Hashim Aljanabi, A.A.; Oxenkrug, G.; Al-Hakeim, H.K.; Maes, M. The Tryptophan Catabolite or Kynurenine Pathway in Major Depressive and Bipolar Disorder: A Systematic Review and Meta-Analysis. Brain Behav. Immun. Health 2022, 26, 100537. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ray, B.; Neavin, D.R.; Zhang, J.; Athreya, A.P.; Biernacka, J.M.; Bobo, W.V.; Hall-Flavin, D.K.; Skime, M.K.; Zhu, H.; et al. Beta-Defensin 1, Aryl Hydrocarbon Receptor and Plasma Kynurenine in Major Depressive Disorder: Metabolomics-Informed Genomics. Transl. Psychiatry 2018, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Setoyama, D.; Kato, T.A.; Hashimoto, R.; Kunugi, H.; Hattori, K.; Hayakawa, K.; Sato-Kasai, M.; Shimokawa, N.; Kaneko, S.; Yoshida, S.; et al. Plasma Metabolites Predict Severity of Depression and Suicidal Ideation in Psychiatric Patients-A Multicenter Pilot Analysis. PLoS ONE 2016, 11, e0165267. [Google Scholar] [CrossRef] [PubMed]
- Messaoud, A.; Rym, M.; Wahiba, D.; Neffati, F.; Najjar, M.F.; Gobbi, G.; Manchia, M.; Valtorta, F.; Lotfi, G.; Comai, S. Investigation of the Relationship among Cortisol, Pro-Inflammatory Cytokines, and the Degradation of Tryptophan into Kynurenine in Patients with Major Depression and Suicidal Behavior. Curr. Top. Med. Chem. 2022, 22, 2119–2125. [Google Scholar] [CrossRef]
- Salimi Elizei, S.; Poormasjedi-Meibod, M.-S.; Wang, X.; Kheirandish, M.; Ghahary, A. Kynurenic Acid Downregulates IL-17/1L-23 Axis in Vitro. Mol. Cell Biochem. 2017, 431, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Baricza, E.; Tamási, V.; Marton, N.; Buzás, E.I.; Nagy, G. The Emerging Role of Aryl Hydrocarbon Receptor in the Activation and Differentiation of Th17 Cells. Cell. Mol. Life Sci. 2016, 73, 95–117. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Rubí, B.; Maechler, P. New Roles for Peripheral Dopamine on Metabolic Control and Tumor Growth: Let’s Seek the Balance. Endocrinology 2010, 151, 5570–5581. [Google Scholar] [CrossRef]
- Fidianingsih, I.; Nurmasitoh, T.; Arjana, A.Z.; Devita, N.; Khoiriyah, U. Mild Anxiety and Depression Related to Elevated Dopamine Level. Universa Med. 2019, 38, 48–55. [Google Scholar] [CrossRef]
- Mills, J.G.; Thomas, S.J.; Larkin, T.A.; Deng, C. Overeating and Food Addiction in Major Depressive Disorder: Links to Peripheral Dopamine. Appetite 2020, 148, 104586. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-X.; Xia, J.-J.; Deng, F.-L.; Liang, W.-W.; Wu, J.; Yin, B.-M.; Dong, M.-X.; Chen, J.-J.; Ye, F.; Wang, H.-Y.; et al. Diagnosis of Major Depressive Disorder Based on Changes in Multiple Plasma Neurotransmitters: A Targeted Metabolomics Study. Transl. Psychiatry 2018, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.B.; Barros, P.O.; Teixeira, B.; Cassano, T.; Centurião, N.; Kasahara, T.M.; Hygino, J.; Vasconcelos, C.C.F.; Filho, H.A.; Alvarenga, R.; et al. Dopamine Favors Expansion of Glucocorticoid-Resistant IL-17-Producing T Cells in Multiple Sclerosis. Brain Behav. Immun. 2014, 41, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Liberman, A.C.; Budziñski, M.L.; Sokn, C.; Gobbini, R.P.; Steininger, A.; Arzt, E. Regulatory and Mechanistic Actions of Glucocorticoids on T and Inflammatory Cells. Front. Endocrinol. 2018, 9, 235. [Google Scholar] [CrossRef]
- Zhao, J.; Lloyd, C.M.; Noble, A. Th17 Responses in Chronic Allergic Airway Inflammation Abrogate Regulatory T-Cell-Mediated Tolerance and Contribute to Airway Remodeling. Mucosal Immunol. 2013, 6, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Tello, A.; Halwani, R.; Hamid, Q.; Al-Muhsen, S. Glucocorticoid Receptor-Beta Up-Regulation and Steroid Resistance Induction by IL-17 and IL-23 Cytokine Stimulation in Peripheral Mononuclear Cells. J. Clin. Immunol. 2013, 33, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-Y.; Wang, C.-H.; Wang, C.-W.; Chen, C.-J.; Huang, H.-Y.; Chung, F.-T.; Huang, Y.-C.; Lin, C.-W.; Lee, C.-S.; Lin, C.-Y.; et al. Increased Interleukin-17 and Glucocorticoid Receptor-β Expression in Interstitial Lung Diseases and Corticosteroid Insensitivity. Front. Immunol. 2022, 13, 905727. [Google Scholar] [CrossRef]
- Al Heialy, S.; Gaudet, M.; Ramakrishnan, R.K.; Mogas, A.; Salameh, L.; Mahboub, B.; Hamid, Q. Contribution of IL-17 in Steroid Hyporesponsiveness in Obese Asthmatics Through Dysregulation of Glucocorticoid Receptors α and β. Front. Immunol. 2020, 11, 1724. [Google Scholar] [CrossRef]
- Xie, H.; Huang, Z.; Wang, R.; Sun, Z. Regulation of Thymocyte Survival by Transcriptional Coactivators. Crit. Rev. Immunol. 2006, 26, 475–486. [Google Scholar] [CrossRef]
- Eberl, G.; Littman, D.R. The Role of the Nuclear Hormone Receptor RORγt in the Development of Lymph Nodes and Peyer’s Patches. Immunol. Rev. 2003, 195, 81–90. [Google Scholar] [CrossRef]
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR Interaction Code: Predictive Value of Direct/Indirect DNA Recruitment for Transcription Outcome. Mol. Cell 2012, 47, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Bulygin, A.S.; Khantakova, J.N.; Shkaruba, N.S.; Shiku, H.; Sennikov, S.S. The Role of Metabolism on Regulatory T Cell Development and Its Impact in Tumor and Transplantation Immunity. Front. Immunol. 2022, 13, 1016670. [Google Scholar] [CrossRef]
- Gowda, B.; Sar, M.; Mu, X.; Cidlowski, J.; Welbourne, T. Coordinate Modulation of Glucocorticoid Receptor and Glutaminase Gene Expression in LLC-PK1-F+ Cells. Am. J. Physiol. -Cell Physiol. 1996, 270, C825–C831. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [PubMed]
- Dankers, W.; Northcott, M.; Bennett, T.; D’Cruz, A.; Sherlock, R.; Gearing, L.J.; Hertzog, P.; Russ, B.; Miceli, I.; Scheer, S.; et al. Type 1 Interferon Suppresses Expression and Glucocorticoid Induction of Glucocorticoid-Induced Leucine Zipper (GILZ). Front. Immunol. 2022, 13, 1034880. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.B.; Hygino, J.; Barros, P.O.; Teixeira, B.; Kasahara, T.M.; Linhares, U.C.; Lopes, L.M.F.; Vasconcelos, C.C.F.; Alvarenga, R.; Wing, A.C.; et al. Endogenous Interleukin-6 Amplifies Interleukin-17 Production and Corticoid-Resistance in Peripheral T Cells from Patients with Multiple Sclerosis. Immunology 2014, 143, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Rahmawati, S.F.; Vos, R.; Bos, I.S.T.; Kerstjens, H.A.M.; Kistemaker, L.E.M.; Gosens, R. Function-Specific IL-17A and Dexamethasone Interactions in Primary Human Airway Epithelial Cells. Sci. Rep. 2022, 12, 11110. [Google Scholar] [CrossRef]
- Nanzer, A.M.; Chambers, E.S.; Ryanna, K.; Richards, D.F.; Black, C.; Timms, P.M.; Martineau, A.R.; Griffiths, C.J.; Corrigan, C.J.; Hawrylowicz, C.M. Enhanced Production of IL-17A in Patients with Severe Asthma Is Inhibited by 1α,25-Dihydroxyvitamin D3 in a Glucocorticoid-Independent Fashion. J. Allergy Clin. Immunol. 2013, 132, 297–304.e3. [Google Scholar] [CrossRef]
- Gupta, A.; Dimeloe, S.; Richards, D.F.; Chambers, E.S.; Black, C.; Urry, Z.; Ryanna, K.; Xystrakis, E.; Bush, A.; Saglani, S.; et al. Defective IL-10 Expression and in Vitro Steroid-Induced IL-17A in Paediatric Severe Therapy-Resistant Asthma. Thorax 2014, 69, 508–515. [Google Scholar] [CrossRef]
- Ouyang, S.; Liu, C.; Xiao, J.; Chen, X.; Lui, A.C.; Li, X. Targeting IL-17A/Glucocorticoid Synergy to CSF3 Expression in Neutrophilic Airway Diseases. JCI Insight 2020, 5, e132836. [Google Scholar] [CrossRef]
- Zijlstra, G.J.; ten Hacken, N.H.T.; Hoffmann, R.F.; van Oosterhout, A.J.M.; Heijink, I.H. Interleukin-17A Induces Glucocorticoid Insensitivity in Human Bronchial Epithelial Cells. Eur. Respir. J. 2012, 39, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Tello, A.; Semlali, A.; Chakir, J.; Martin, J.G.; Leung, D.Y.; Eidelman, D.H.; Hamid, Q. Induction of Glucocorticoid Receptor-β Expression in Epithelial Cells of Asthmatic Airways by T-Helper Type 17 Cytokines. Clin. Exp. Allergy 2010, 40, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Halwani, R.; Sultana, A.; Al-Kufaidy, R.; Jamhawi, A.; Vazquez-Tello, A.; Al-Muhsen, S. Th-17 Regulatory Cytokines Inhibit Corticosteroid Induced Airway Structural Cells Apoptosis. Respir Res 2016, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, C.; Kaphalia, L.; Calhoun, W.J. The Effect of IL-17 on Glucocorticoid Receptor [GR] Translocation. J. Allergy Clin. Immunol. 2011, 127, AB125. [Google Scholar] [CrossRef]
- Vincken, N.L.A.; Balak, D.M.W.; Knulst, A.C.; Welsing, P.M.J.; van Laar, J.M. Systemic Glucocorticoid Use and the Occurrence of Flares in Psoriatic Arthritis and Psoriasis: A Systematic Review. Rheumatology 2022, 61, 4232–4244. [Google Scholar] [CrossRef] [PubMed]
- Le Page, E.; Veillard, D.; Laplaud, D.A.; Hamonic, S.; Wardi, R.; Lebrun, C.; Zagnoli, F.; Wiertlewski, S.; Deburghgraeve, V.; Coustans, M.; et al. Oral versus Intravenous High-Dose Methylprednisolone for Treatment of Relapses in Patients with Multiple Sclerosis (COPOUSEP): A Randomised, Controlled, Double-Blind, Non-Inferiority Trial. Lancet 2015, 386, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Ora, J.; Calzetta, L.; Matera, M.G.; Cazzola, M.; Rogliani, P. Advances with Glucocorticoids in the Treatment of Asthma: State of the Art. Expert Opin. Pharmacother. 2020, 21, 2305–2316. [Google Scholar] [CrossRef]
- Tsujimura, S.; Saito, K.; Nawata, M.; Nakayamada, S.; Tanaka, Y. Overcoming Drug Resistance Induced by P-Glycoprotein on Lymphocytes in Patients with Refractory Rheumatoid Arthritis. Ann. Rheum. Dis. 2007, 67, 380–388. [Google Scholar] [CrossRef]
- Iqbal, M.; Baello, S.; Javam, M.; Audette, M.C.; Gibb, W.; Matthews, S.G. Regulation of Multidrug Resistance P-Glycoprotein in the Developing Blood-Brain Barrier: Interplay between Glucocorticoids and Cytokines. J. Neuroendocrinol. 2016, 28, 12360. [Google Scholar] [CrossRef]
- De Iudicibus, S. Molecular Mechanism of Glucocorticoid Resistance in Inflammatory Bowel Disease. World J. Gastroenterol. 2011, 17, 1095. [Google Scholar] [CrossRef]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-Inflammatory Human Th17 Cells Selectively Express P-Glycoprotein and Are Refractory to Glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Koetzier, S.C.; van Langelaar, J.; Blok, K.M.; van den Bosch, T.P.P.; Wierenga-Wolf, A.F.; Melief, M.-J.; Pol, K.; Siepman, T.A.; Verjans, G.M.G.M.; Smolders, J.; et al. Brain-Homing CD4+ T Cells Display Glucocorticoid-Resistant Features in MS. Neurol. Neuroimmunol. Neuroinflammation 2020, 7, e894. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Non-Pathogenic Th17 | Pathogenic Th17 | |
|---|---|---|
| Factor of differentiation | TGF-β, IL-6 | TGF-β, IL-6, IL-23 + IL-1β, IL-12, TNF |
| Transcription factors | RORγt, STAT3, c-Maf, AHR, Ets1, Batch2, Fosl2, Rbpj | RORγt, STAT3, Atf3, Bhlhe40, Fos, Nr4a1, Eomes, Tbx21 |
| Surface receptors | CCR4, CCR6, IL6R, CD5L | IL-23R, CCR4, CCR6, IL6R, CXCR3 |
| Cytokines | IL-10, IL-9, IL-17, IL-21 | IL-17, IL-21, IL-22, GM-CSF, IFNγ, Granzime B |
| Metabolism | Oxidative phosphorylation | Glycolysis |
| Function |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khantakova, J.N.; Mutovina, A.; Ayriyants, K.A.; Bondar, N.P. Th17 Cells, Glucocorticoid Resistance, and Depression. Cells 2023, 12, 2749. https://doi.org/10.3390/cells12232749
Khantakova JN, Mutovina A, Ayriyants KA, Bondar NP. Th17 Cells, Glucocorticoid Resistance, and Depression. Cells. 2023; 12(23):2749. https://doi.org/10.3390/cells12232749
Chicago/Turabian StyleKhantakova, Julia N., Anastasia Mutovina, Kseniya A. Ayriyants, and Natalia P. Bondar. 2023. "Th17 Cells, Glucocorticoid Resistance, and Depression" Cells 12, no. 23: 2749. https://doi.org/10.3390/cells12232749