
Histopathological Markers for Target Therapies in Primary Cutaneous Lymphomas
 , ,
, ,
Abstract
:
1. Introduction
2. Principles of Histopathology, Immunohistochemistry, and Biomarkers of CTCL and BPDCN
2.1. Histopathology of the Patch-Plaque Stage of Mycosis Fungoides
2.2. Immunohistochemistry of Mycosis Fungoides
2.3. Histopathology of Tumor Stage of Mycosis Fungoides and CD30 Antigen
2.4. Histopathology and Immunohistochemistry of Lymphomatoid Papulosis
2.5. Histopathology and Immunohistochemistry of Primary Cutaneous Anaplastic Large T-Cell Lymphoma
2.6. Histopathology and Immunohistochemistry of Sezary Syndrome
2.7. Histopathology and Immunohistochemistry of Blastic Plasmacytoid Dendritic Cell Neoplasm
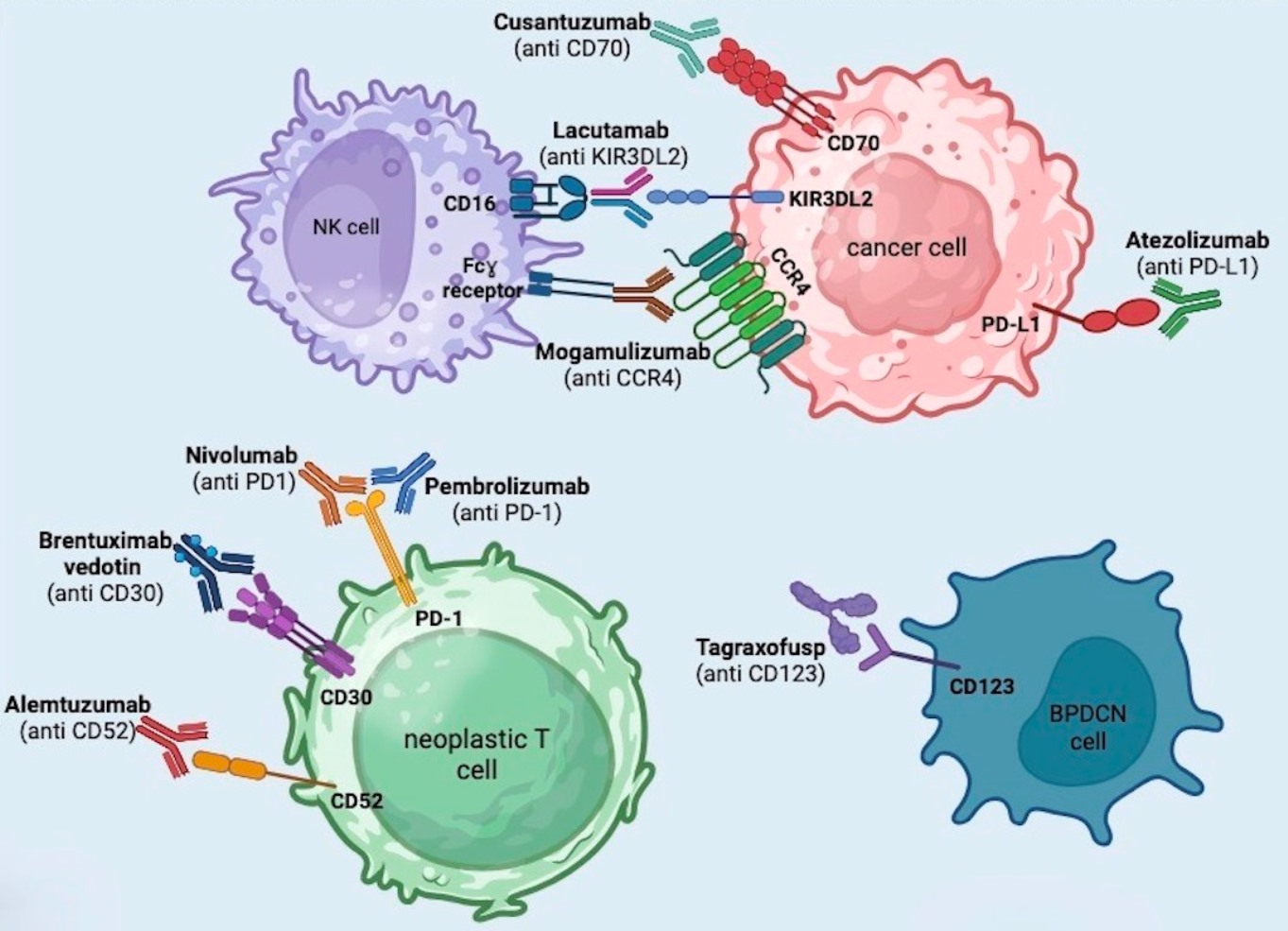
3. Brentuximab Vedotin
4. Mogamulizumab
5. Anti-PD1 and Anti-PDL1 (Pembrolizumab + Nivolumab + Atezolizumab)
6. Alemtuzumab
7. Lacutamab
8. Cusatuzumab
9. Tagraxofusp
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714, Erratum in Blood 2019, 134, 1112. [Google Scholar] [CrossRef]
- Willemze, R.; Jansen, P.M.; Cerroni, L.; Berti, E.; Santucci, M.; Assaf, C.; Canninga-van Dijk, M.R.; Carlotti, A.; Geerts, M.L.; Hahtola, S.; et al. Subcutaneous panniculitis-like T-cell lymphoma: Definition, classification, and prognostic factors: An EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008, 111, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R. Primary cutaneous lymphoma: The 2018 update of the WHO-EORTC classification. Presse Med. 2022, 51, 104126. [Google Scholar] [CrossRef] [PubMed]
- Alberti Violetti, S.; Alaibac, M.; Ardigò, M.; Baldo, A.; DIMeo, N.; Massone, C.; Onida, F.; Simontacchi, G.; Zalaudek, I.; Pimpinelli, N.; et al. An expert consensus report on mycosis fungoides in Italy: Epidemiological impact and diagnostic-therapeutic pathway. Ital. J. Dermatol. Venerol. 2021, 156, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Massone, C.; Chott, A.; Metze, D.; Kerl, K.; Citarella, L.; Vale, E.; Kerl, H.; Cerroni, L. Subcutaneous, blastic natural killer (NK), NK/T-cell, and other cytotoxic lymphomas of the skin: A morphologic, immunophenotypic, and molecular study of 50 patients. Am. J. Surg. Pathol. 2004, 28, 719–735. [Google Scholar] [CrossRef]
- Cota, C.; Vale, E.; Viana, I.; Requena, L.; Ferrara, G.; Anemona, L.; Metze, D.; Fink-Puches, R.; Wiesner, T.; Cerroni, L. Cutaneous manifestations of blastic plasmacytoid dendritic cell neoplasm-morphologic and phenotypic variability in a series of 33 patients. Am. J. Surg. Pathol. 2010, 34, 75–87. [Google Scholar] [CrossRef]
- Pagano, L.; Zinzani, P.L.; Pileri, S.; Quaglino, P.; Cuglievan, B.; Berti, E.; Pemmaraju, N.; Onida, F.; Willemze, R.; Orfao, A.; et al. Unmet Clinical Needs and Management Recommendations for Blastic Plasmacytoid Dendritic Cell Neoplasm: A Consensus-based Position Paper from an Ad Hoc International Expert Panel. Hemasphere 2023, 7, e841. [Google Scholar] [CrossRef]
- Laribi, K.; Baugier de Materre, A.; Sobh, M.; Cerroni, L.; Valentini, C.G.; Aoki, T.; Suzuki, R.; Takeuchi, K.; Frankel, A.E.; Cota, C.; et al. Blastic plasmacytoid dendritic cell neoplasms: Results of an international survey on 398 adult patients. Blood Adv. 2020, 4, 4838–4848. [Google Scholar] [CrossRef]
- Facchetti, F.; Jones, D.; Petrella, T. Blastic plasmacytoid dendritic cell neoplasm. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; IARC Press: Lyon, France, 2008; pp. 145–147. [Google Scholar]
- Petrella, T.; Dalac, S.; Maynadie, M.; Mugneret, F.; Thomine, E.; Courville, P.; Joly, P.; Lenormand, B.; Arnould, L.; Wechsler, J.; et al. CD4 + CD56 + cutaneous neoplasms: A distinct hematological entity? Groupe Francais d’Etude des Lymphomes Cutanes (GFELC). Am. J. Surg. Pathol. 1999, 23, 137–146. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Kantarjian, H. Blastic plasmacytoid dendritic cell neoplasm: Emerging developments and special considerations for 2023. Clin. Adv. Hematol. Oncol. 2023, 21, 257–264. [Google Scholar]
- Fava, P.; Roccuzzo, G.; Alberti-Violetti, S.; Grandi, V.; Pileri, A.; Pimpinelli, N.; Berti, E.; Quaglino, P. Cutaneous B-cell lymphomas: Update on diagnosis, risk-stratification, and management. Presse Med. 2022, 51, 104109. [Google Scholar] [CrossRef] [PubMed]
- Trautinger, F.; Eder, J.; Assaf, C.; Bagot, M.; Cozzio, A.; Dummer, R.; Gniadecki, R.; Klemke, C.D.; Ortiz-Romero, P.L.; Papadavid, E.; et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—Update 2017. Eur. J. Cancer 2017, 77, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.M.; Scarisbrick, J.J.; Dummer, R.; Whittaker, S.; Duvic, M.; Kim, Y.H.; Quaglino, P.; Zinzani, P.L.; Bechter, O.; Eradat, H.; et al. Randomized phase 3 ALCANZA study of brentuximab vedotin vs physician’s choice in cutaneous T-cell lymphoma: Final data. Blood Adv. 2021, 5, 5098–5106. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, P.; Fava, P.; Pileri, A.; Grandi, V.; Sanlorenzo, M.; Panasiti, V.; Guglielmo, A.; Alberti-Violetti, S.; Novelli, M.; Astrua, C.; et al. Phenotypical Markers, Molecular Mutations, and Immune Microenvironment as Targets for New Treatments in Patients with Mycosis Fungoides and/or Sézary Syndrome. J. Invest. Dermatol. 2021, 141, 484–495. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Quaglino, P.; Violetti, S.A.; Cantonetti, M.; Goteri, G.; Onida, F.; Paulli, M.; Rupoli, S.; Barosi, G.; Pimpinelli, N. Critical concepts and management recommendations for cutaneous T-cell lymphoma: A consensus-based position paper from the Italian Group of Cutaneous Lymphoma. Hematol. Oncol. 2021, 39, 275–283. [Google Scholar] [CrossRef]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Afifi, S.; Mohamed, S.; Zhao, J.; Foss, F. A drug safety evaluation of mogamulizumab for the treatment of cutaneous T-Cell lymphoma. Expert. Opin. Drug Saf. 2019, 18, 769–776. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sézary Syndrome: A Multicenter Phase II Study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef]
- de Masson, A. New biotherapies for the treatment of cutaneous T-cell lymphomas. Presse Med. 2022, 51, 104110. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Stadler, R.; Romero, P.O.; Bagot, M.; Quaglino, P.; Guenova, E.; Jonak, C.; Papadavid, E.; Stranzenbach, R.; Sartori, D.; Musoro, J.Z.; et al. Phase II trial of atezolizumab (anti-PD-L1) in the treatment of stage IIb-IVB mycosis fungoides/Sézary syndrome patients relapsed/refractory after a previous systemic treatment (PARCT). Eur. J. Cancer. 2021, 156, S22–S23. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Mehta, N.; Rosen, S.T.; Guitart, J.; Rademaker, A.; Gerami, P. Alemtuzumab for relapsed and refractory erythrodermic cutaneous T-cell lymphoma: A single institution experience from the Robert H. Lurie Comprehensive Cancer Center. Leuk. Lymphoma 2009, 50, 1969–1976. [Google Scholar] [CrossRef] [PubMed]
- de Masson, A.; Guitera, P.; Brice, P.; Moulonguet, I.; Mouly, F.; Bouaziz, J.D.; Battistella, M.; Madelaine, I.; Roux, J.; Ram-Wolff, C.; et al. Long-term efficacy and safety of alemtuzumab in advanced primary cutaneous T-cell lymphomas. Br. J. Dermatol. 2014, 170, 720–724. [Google Scholar] [CrossRef]
- Bagot, M.; Porcu, P.; Marie-Cardine, A.; Battistella, M.; William, B.M.; Vermeer, M.; Whittaker, S.; Rotolo, F.; Ram-Wolff, C.; Khodadoust, M.S.; et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: An international, first-inhuman, open-label, phase 1 trial. Lancet Oncol. 2019, 20, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Leupin, N.; Zinzani, P.L.; Morschhauser, F.; Dalle, S.; Maerevoet, M.; Michot, J.M.; Ribrag, V.; Offner, F.; Beylot-Barry, M.; Moins-Teisserenc, H.; et al. Cusatuzumab for treatment of CD70-positive relapsed or refractory cutaneous T-cell lymphoma. Cancer 2022, 128, 1004–1014. [Google Scholar] [CrossRef]
- Moretti, S.; Lanza, F.; Dabusti, M.; Tieghi, A.; Campioni, D.; Dominici, M.; Castoldi, G.L. CD123 (interleukin 3 receptor alpha chain). J. Biol. Regul. Homeost. Agents. 2001, 15, 98–100. [Google Scholar]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef]
- Massone, C.; Kodama, K.; Kerl, H.; Cerroni, L. Histopathologic features of early (patch) lesions of mycosis fungoides: A morphologic study on 745 biopsy specimens from 427 patients. Am. J. Surg. Pathol. 2005, 29, 550–560. [Google Scholar] [CrossRef]
- Pimpinelli, N.; Olsen, E.A.; Santucci, M.; Vonderheid, E.; Haeffner, A.C.; Stevens, S.; Burg, G.; Cerroni, L.; Dreno, B.; Glusac, E.; et al. Defining early mycosis fungoides. J. Am. Acad. Dermatol. 2005, 53, 1053–1063. [Google Scholar] [CrossRef]
- Cerroni, L. Skin Lymphoma, The Illustrated Guide, 5th ed.; Wiley-Blackwell: London, UK, 2020. [Google Scholar]
- Massone, C.; Crisman, G.; Kerl, H.; Cerroni, L. The prognosis of early mycosis fungoides is not influenced by phenotype and T-cell clonality. Br. J. Dermatol. 2008, 159, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Kash, N.; Massone, C.; Fink-Puches, R.; Cerroni, L. Phenotypic Variation in Different Lesions of Mycosis Fungoides Biopsied Within a Short Period of Time from the Same Patient. Am. J. Dermatopathol. 2016, 38, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Alberti-Violetti, S.; Sapienza, M.R.; Del Corvo, M.; Melle, F.; Motta, G.; Venegoni, L.; Cerroni, L.; Cota, C.; Pileri, A.; Berti, E.; et al. A Microenvironment-Related Nine-Gene Signature May Predict Survival in Mycosis Fungoides Patients at Diagnosis. Cells 2023, 12, 1944. [Google Scholar] [CrossRef]
- Geller, S.; Hollmann, T.J.; Horwitz, S.M.; Myskowski, P.L.; Pulitzer, M. C-C chemokine receptor 4 expression in CD8+ cutaneous T-cell lymphomas and lymphoproliferative disorders, and its implications for diagnosis and treatment. Histopathology 2020, 76, 222–232. [Google Scholar] [CrossRef]
- Ahn, C.S.; ALSayyah, A.; Sangüeza, O.P. Mycosis fungoides: An updated review of clinicopathologic variants. Am. J. Dermatopathol. 2014, 36, 933–948; quiz 949–951. [Google Scholar] [CrossRef]
- Reiter, O.; Amitay-Laish, I.; Oren-Shabtai, M.; Feinmesser, M.; Ben-Amitai, D.; Hodak, E. Paediatric mycosis fungoides—Characteristics, management and outcomes with particular focus on the folliculotropic variant. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 671–679, Erratum in J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1143. [Google Scholar] [CrossRef]
- Edinger, J.T.; Clark, B.Z.; Pucevich, B.E.; Geskin, L.J.; Swerdlow, S.H. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am. J. Surg. Pathol. 2009, 33, 1860–1868. [Google Scholar] [CrossRef]
- Kempf, W.; Mitteldorf, C. Cutaneous T-cell lymphomas—An update 2021. Hematol Oncol. 2021, 39, 46–51. [Google Scholar] [CrossRef]
- Werner, B.; Massone, C.; Kerl, H.; Cerroni, L. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J. Cutan. Pathol. 2008, 35, 1100–1107. [Google Scholar] [CrossRef]
- Wehkamp, U.; Mitteldorf, C.; Stendel, S.; Stranzenbach, R.; Nicolay, J.P.; Wobser, M.; Weichenthal, M.; Schneiderbauer, R.; Klemke, C.D.; Hillen, U.; et al. Most rare subtypes of cutaneous lymphoma display variable CD30 expression: Analysis of the German Cutaneous Lymphoma Network. Br. J. Dermatol. 2021, 185, 228–230. [Google Scholar] [CrossRef]
- Kempf, W. A new era for cutaneous CD30-positive T-cell lymphoproliferative disorders. Semin. Diagn. Pathol. 2017, 34, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Massone, C.; El-Shabrawi-Caelen, L.; Kerl, H.; Cerroni, L. The morphologic spectrum of primary cutaneous anaplastic large T-cell lymphoma: A histopathologic study on 66 biopsy specimens from 47 patients with report of rare variants. J. Cutan. Pathol. 2008, 35, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Massone, C.; Cerroni, L. Phenotypic variability in primary cutaneous anaplastic large T-cell lymphoma: A study on 35 patients. Am. J. Dermatopathol. 2014, 36, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jiang, Y.; Xu, X.; Sun, J.; Chen, H. Pediatric Primary Cutaneous Lymphoma in China: A Retrospective Study. Dermatology 2023, 239, 553–564. [Google Scholar] [CrossRef]
- Niu, N.; Heberton, M.M.; Tang, Z.; Aung, P.P.; Nagarajan, P.; Curry, J.L.; Prieto, V.G.; Torres-Cabala, C.A.; Cho, W.C. Lymphomatoid papulosis with DUSP22-IRF4 rearrangement: A case report and literature review. J. Cutan. Pathol. 2023, 50, 711–716. [Google Scholar] [CrossRef]
- Nowicka, D.; Mertowska, P.; Mertowski, S.; Hymos, A.; Forma, A.; Michalski, A.; Morawska, I.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Grywalska, E. Etiopathogenesis, Diagnosis, and Treatment Strategies for Lymphomatoid Papulosis with Particular Emphasis on the Role of the Immune System. Cells 2022, 11, 3697. [Google Scholar] [CrossRef]
- Pileri, A.; Tabanelli, V.; Fuligni, F.; Agostinelli, C.; Guglielmo, A.; Sabattini, E.; Grandi, V.; Pileri, S.A.; Pimpinelli, N. PD-1 and PD-L1 expression in mycosis fungoides and Sézary Syndrome. Ital. J. Dermatol. Venerol. 2022, 157, 355–362. [Google Scholar] [CrossRef]
- Di Raimondo, C.; Rubio-Gonzalez, B.; Palmer, J.; Weisenburger, D.D.; Zain, J.; Wu, X.; Han, Z.; Rosen, S.T.; Song, J.Y.; Querfeld, C. Expression of immune checkpoint molecules programmed death protein 1, programmed death-ligand 1 and inducible T-cell co-stimulator in mycosis fungoides and Sézary syndrome: Association with disease stage and clinical outcome. Br. J. Dermatol. 2022, 187, 234–243. [Google Scholar] [CrossRef]
- Ascani, S.; Massone, C.; Ferrara, G.; Rongioletti, F.; Papini, M.; Pileri, S.; Cerroni, L. CD4-negative variant of CD4+/CD56+ hematodermic neoplasm: Description of three cases. J. Cutan. Pathol. 2008, 35, 911–915. [Google Scholar] [CrossRef]
- Massone, C.; Raiola, A.M.; Dominietto, A.; Minetto, P.; Beltramini, S.; Cerroni, L.; Sola, S.; Angelucci, E. Blastic Plasmacytoid Dendritic Cell Neoplasm: Underlining the importance of an early diagnosis and the use of tagraxofusp therapy before wide dissemination. Australas. J. Dermatol. 2021, 62, e316–e318. [Google Scholar] [CrossRef]
- Kim, Y.H.; Prince, H.M.; Whittaker, S.; Horwitz, S.M.; Duvic, M.; Bechter, O.; Sanches, J.A.; Stadler, R.; Scarisbrick, J.; Quaglino, P.; et al. Response to brentuximab vedotin versus physician’s choice by CD30 expression and large cell transformation status in patients with mycosis fungoides: An ALCANZA sub-analysis. Eur. J. Cancer. 2021, 148, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Mou, E.; Kim, Y.H. Integrating novel agents into the treatment of advanced mycosis fungoides and Sézary syndrome. Blood 2023, 141, 695–703. [Google Scholar] [CrossRef]
- Scarisbrick, J.J. Brentuximab vedotin therapy for CD30-positive cutaneous T-cell lymphoma: A targeted approach to management. Future Oncol. 2017, 13, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Corbin, Z.A.; Nguyen-Lin, A.; Li, S.; Rahbar, Z.; Tavallaee, M.; Vogel, H.; Salva, K.A.; Wood, G.S.; Kim, Y.H.; Nagpal, S. Characterization of the peripheral neuropathy associated with brentuximab vedotin treatment of Mycosis Fungoides and Sézary Syndrome. J. Neurooncol. 2017, 132, 439–446. [Google Scholar] [CrossRef] [PubMed]
- André, R.; Ram-Wolff, C.; Battistella, M.; Peffault de Latour, R.; Petit, A.; Bouaziz, J.D.; Brice, P.; Bagot, M.; de Masson, A. Effectiveness of brentuximab vedotin before and after allogeneic stem-cell transplantation in the management of transformed mycosis fungoides. Br. J. Dermatol. 2020, 182, 1503–1504. [Google Scholar] [CrossRef]
- Bogacka, J.; Pawlik, K.; Ciapała, K.; Ciechanowska, A.; Mika, J. CC Chemokine Receptor 4 (CCR4) as a Possible New Target for Therapy. Int. J. Mol. Sci. 2022, 23, 15638. [Google Scholar] [CrossRef]
- Blackmon, A.L.; Pinter-Brown, L. Spotlight on Mogamulizumab-Kpkc for Use in Adults with Relapsed or Refractory Mycosis Fungoides or Sézary Syndrome: Efficacy, Safety, and Patient Selection. Drug Des. Devel. Ther. 2020, 14, 3747–3754. [Google Scholar] [CrossRef]
- Cowan, R.A.; Scarisbrick, J.J.; Zinzani, P.L.; Nicolay, J.P.; Sokol, L.; Pinter-Brown, L.; Quaglino, P.; Iversen, L.; Dummer, R.; Musiek, A.; et al. Efficacy and safety of mogamulizumab by patient baseline blood tumour burden: A post hoc analysis of the MAVORIC trial. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 2225–2238. [Google Scholar] [CrossRef]
- Bonnet, P.; Battistella, M.; Roelens, M.; Ram-Wolff, C.; Herms, F.; Frumholtz, L.; Bouaziz, J.D.; Brice, P.; Moins-Teisserenc, H.; Bagot, M.; et al. Association of autoimmunity and long-term complete remission in patients with Sezary syndrome treated with mogamulizumab. Br. J. Dermatol. 2019, 180, 419–420. [Google Scholar] [CrossRef]
- Trum, N.A.; Zain, J.; Martinez, X.U.; Parekh, V.; Afkhami, M.; Abdulla, F.; Carson, K.R.; Rosen, S.T.; Bennett, C.L.; Querfeld, C. Mogamulizumab efficacy is underscored by its associated rash that mimics cutaneous T-cell lymphoma: A retrospective single-centre case series. Br. J. Dermatol. 2022, 186, 153–166. [Google Scholar] [CrossRef]
- Musiek, A.C.M.; Rieger, K.E.; Bagot, M.; Choi, J.N.; Fisher, D.C.; Guitart, J.; Haun, P.L.; Horwitz, S.M.; Huen, A.O.; Kwong, B.Y.; et al. Dermatologic Events Associated with the Anti-CCR4 Antibody Mogamulizumab: Characterization and Management. Dermatol. Ther. 2022, 12, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, K.E.; Neal, T.M.; Khodadoust, M.S.; Wang, J.Y.; Rieger, K.E.; Strelo, J.; Hong, E.; Kim, Y.H.; Kwong, B.Y. Clinical Characterization of Mogamulizumab-Associated Rash During Treatment of Mycosis Fungoides or Sézary Syndrome. JAMA Dermatol. 2021, 157, 700–707. [Google Scholar] [CrossRef] [PubMed]
- de Masson, A.; Darbord, D.; Dobos, G.; Boisson, M.; Roelens, M.; Ram-Wolff, C.; Cassius, C.; Le Buanec, H.; de la Grange, P.; Jouenne, F.; et al. Macrophage-derived CXCL9 and CXCL11, T-cell skin homing, and disease control in mogamulizumab-treated CTCL patients. Blood 2022, 139, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Pileri, A.; Clarizio, G.; Zengarini, C.; Casadei, B.; Sabattini, E.; Agostinelli, C.; Zinzani, P.L. Mogamulizumab-associated rashes, their presentation and prognostic significance: A single-centre retrospective case series analysis. J. Eur. Acad. Dermatol. Venereol. 2023, 37, e615–e617. [Google Scholar] [CrossRef]
- Jfri, A.; Virgen, C.A.; Tawa, M.; Giobbie-Hurder, A.; Kupper, T.S.; Fisher, D.C.; LeBoeuf, N.R.; Larocca, C. Prevalence and implications of mogamulizumab-induced immune-related adverse events in mycosis fungoides/Sézary syndrome; a single-center experience. J. Am. Acad. Dermatol. 2023, 89, 1044–1047. [Google Scholar] [CrossRef]
- Dai, J.; Almazan, T.H.; Hong, E.K.; Khodadoust, M.S.; Arai, S.; Weng, W.K.; Kim, Y.H. Potential association of anti-CCR4 antibody mogamulizumab and graft-vs-host disease in patients with mycosis Fungoides and Sezary syndrome. JAMA Dermatol. 2018, 154, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Mogamulizumab [Package Insert]; Kyowa Kirin, Inc.: Bedminster, NJ, USA, 2018.
- Jouandet, M.; Nakouri, I.; Nadin, L.; Kieny, A.; Samimi, M.; Adamski, H.; Quéreux, G.; Chaby, G.; Dompmartin, A.; L’Orphelin, J.M. Impact of Mogamulizumab in Real-Life Advanced Cutaneous T-Cell Lymphomas: A Multicentric Retrospective Cohort Study. Cancers 2022, 14, 1659. [Google Scholar] [CrossRef]
- Beylot-Barry, M.; Quereux, G.; Nardin, C.; Duval-Modeste, A.B.; Dereure, O.; Dalac-Rat, S.; Dobos, G.; Pham-Ledard, A.; Ram-Wolff, C.; D’Incan, M.; et al. Effectiveness of mogamulizumab in patients with Mycosis Fungoides or Sézary syndrome: A multicentre, retrospective, real-world French study. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 1777–1784. [Google Scholar] [CrossRef]
- Caruso, L.; Castellino, A.; Dessì, D.; Flenghi, L.; Giordano, A.; Ibatici, A.; Massone, C.; Pileri, A.; Proietti, I.; Pupo, L.; et al. Italian Real-Life Experience on the Use of Mogamulizumab in Patients with Cutaneous T-Cell Lymphomas. Cancer Manag. Res. 2022, 14, 3205–3221. [Google Scholar] [CrossRef]
- Fong, S.; Hong, E.K.; Khodadoust, M.S.; Li, S.; Hoppe, R.T.; Kim, Y.H.; Hiniker, S.M. Low-Dose Total Skin Electron Beam Therapy Combined with Mogamulizumab for Refractory Mycosis Fungoides and Sézary Syndrome. Adv. Radiat. Oncol. 2020, 6, 100629. [Google Scholar] [CrossRef]
- Anti-CCR4 Monoclonal Antibody (Mogamulizumab) and Total Skin Electron Beam Therapy (TSEB) in Patients with Stage IB-IIB Cutaneous T-Cell Lymphoma (MOGAT). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04128072 (accessed on 20 August 2023).
- Porcu, P.; Hudgens, S.; Horwitz, S.; Quaglino, P.; Cowan, R.; Geskin, L.; Beylot-Barry, M.; Floden, L.; Bagot, M.; Tsianakas, A.; et al. Quality of Life Effect of the Anti-CCR4 Monoclonal Antibody Mogamulizumab Versus Vorinostat in Patients with Cutaneous T-cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.; Zinzani, P.L.; Bagot, M.; Kim, Y.H.; Moskowitz, A.J.; Porcu, P.; Dwyer, K.; Sun, W.; Herr, F.M.; Scarisbrick, J. Lack of impact of type and extent of prior therapy on outcomes of mogamulizumab therapy in patients with cutaneous T cell lymphoma in the MAVORIC trial. Leuk. Lymphoma 2021, 62, 3109–3118. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Teague, J.E.; Fisher, D.C.; Kupper, T.S.; Clark, R.A. Alemtuzumab Therapy for Leukemic Cutaneous T-Cell Lymphoma: Diffuse Erythema as a Positive Predictor of Complete Remission. JAMA Dermatol. 2014, 150, 776–779. [Google Scholar] [CrossRef]
- Lundin, J.; Osterborg, A.; Brittinger, G.; Crowther, D.; Dombret, H.; Engert, A.; Epenetos, A.; Gisselbrecht, C.; Huhn, D.; Jaeger, U.; et al. CAMPATH-1H monoclonal antibody in therapy for previously treated low-grade non-Hodgkin’s lymphomas: A phase II multicenter study. European Study Group of CAMPATH-1H Treatment in Low-Grade Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 1998, 16, 3257–3263. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.A.; Seymour, J.F.; Wolf, M.; Januszewicz, H.; Davison, J.; McCormack, C.; Ryan, G.; Prince, H.M. Treatment of patients with advanced mycosis fungoides and Sézary syndrome with alemtuzumab. Eur. J. Haematol. 2003, 71, 250–256. [Google Scholar] [CrossRef]
- Weder, P.; Anliker, M.; Itin, P.; Bargetzi, M. Familial cutaneous mycosis fungoides: Successful treatment with a combination of gemcitabine and alemtuzumab. Dermatology 2004, 208, 281–283. [Google Scholar] [CrossRef]
- Bernengo, M.G.; Quaglino, P.; Comessatti, A.; Ortoncelli, M.; Novelli, M.; Lisa, F.; Fierro, M.T. Low-dose intermittent alemtuzumab in the treatment of Sézary syndrome: Clinical and immunologic findings in 14 patients. Haematologica 2007, 92, 784–794. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Disease | Treatment Schedule | ORR | PFS | References |
|---|---|---|---|---|---|---|
| Brentuximab vedotin | CD-30 | CD30+ CTCL after at least 1 prior systemic therapy | 1.8 mg/kg iv Q3w for 16 cycles | 67% | 16.7 mths | [15,18] |
| Mogamulizumab | CCR-4 | R/R MF and SS | 1.0 mg/kg iv qw for the first 28-days cycle, then on days 1 and 15 of subsequent cycles | 28% | 7.7 mths | [14,19] |
| Pembrolizumab | PD-1 | R/R MF and SS | 2 mg/kg iv Q3w for 24 months | 38% | 65% at 1 year | [20] |
| Nivolumab | PD-1 | R/R CTCL | 1 or 3 mg/kg at weeks 1 and 4, then Q2w for up to 2 years | - | 10 mths | [21,22] |
| Atezolizumab | PD-L1 | R/R stage IIb-IV MF and SS | 1200 mg IV Q3w for 1 year | ongoing study | ongoing study | [23] |
| Alemtuzumab | CD-52 | stage IIb-IV MF and SS | increasing doses from 3–10 mg to 30 mg i.v. q.o.d., followed by 30 mg t.i.w. for at least 12 weeks | 51% | 56 mths | [24,25] |
| Lacutamab | KIR3DL2 | R/R CTCL | increasing doses from 0.0001 mg/kg to 10 mg/kg Q1w for the first month, Q2w for 10 doses, then Q4w | 36.40% | 8.2 mths | [26] |
| Cusantuzumab | CD70-CD27 | R/R CTCL | 1 mg/kg or 5 mg/kg Q3w | 23% | - | [27] |
| Tagraxofusp | CD123 | BPDCN | 12 mcg/kg q.d. on days 1 to 5 of each 21-day cycle | 75% as first-line 58% in R/R patients | - | [28,29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonego, B.; Ibatici, A.; Rivoli, G.; Angelucci, E.; Sola, S.; Massone, C. Histopathological Markers for Target Therapies in Primary Cutaneous Lymphomas. Cells 2023, 12, 2656. https://doi.org/10.3390/cells12222656
Sonego B, Ibatici A, Rivoli G, Angelucci E, Sola S, Massone C. Histopathological Markers for Target Therapies in Primary Cutaneous Lymphomas. Cells. 2023; 12(22):2656. https://doi.org/10.3390/cells12222656
Chicago/Turabian StyleSonego, Benedetta, Adalberto Ibatici, Giulia Rivoli, Emanuele Angelucci, Simona Sola, and Cesare Massone. 2023. "Histopathological Markers for Target Therapies in Primary Cutaneous Lymphomas" Cells 12, no. 22: 2656. https://doi.org/10.3390/cells12222656