
Mechanisms of Endothelial Cell Membrane Repair: Progress and Perspectives

 ,
,
Abstract
:1. Introduction
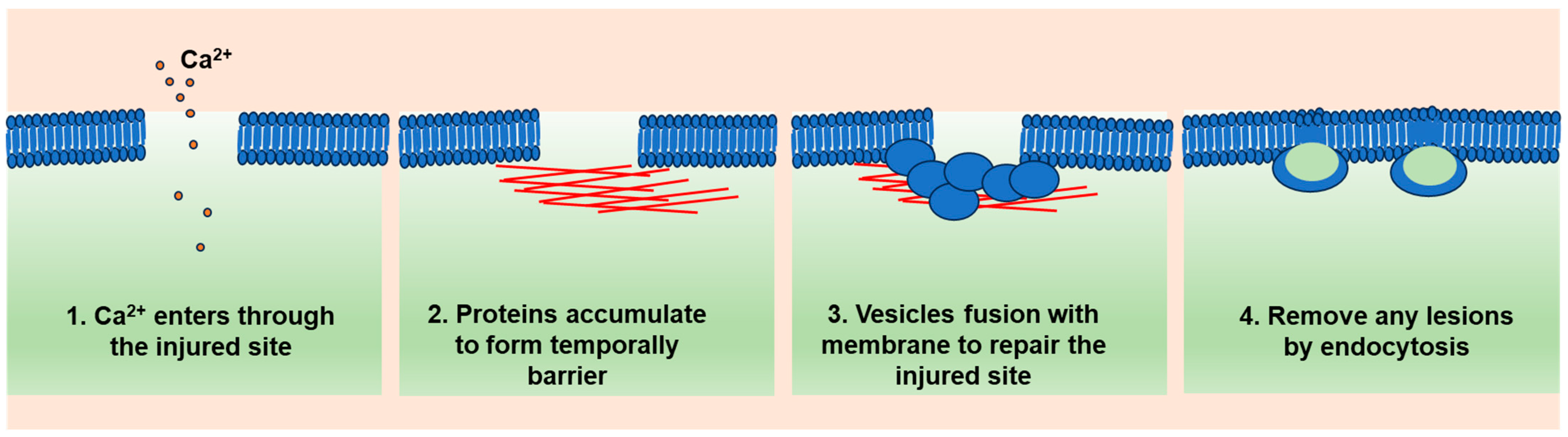
2. A General Model of Cell Membrane Repair
2.1. Ca2+ Influx as a Trigger
2.2. Ca2+-Targeted Proteins Form a Temporary Diffusion Barrier (Resealing Phase)
2.3. Ca2+-Regulated Lysosome Exocytosis (Repairing Phase)
2.4. Lesion Removal by Endocytosis
3. Endothelial Cell Membrane Damage
3.1. Mechanical Stresses
3.2. Chemical Stresses
3.3. Biological Stresses
4. Endothelial Cell Membrane Repair
4.1. Dysferlin and Myoferlin
4.2. Annexins and S100A10/11
4.3. Caveolins
4.4. Weibel–Palade Bodies
4.5. Other Proteins
5. The Diseases Linked with Defective Endothelial Membrane Repair
5.1. Atherosclerosis
5.2. Diabetes Mellitus
5.3. Acute Respiratory Distress Syndrome (ARDS) and Idiopathic Pulmonary Fibrosis (IPF)
6. The Resealing Agents for Endothelial Membrane Repair
6.1. Recombinant MG53
6.2. Recombinant Annexins
6.3. Poloxamer 188
6.4. Acid Sphingomyelinase (ASM), Sphingomyelin, or Ceramide
7. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Yu, Q.C.; McNeil, P.L. Transient disruptions of aortic endothelial cell plasma membranes. Am. J. Pathol. 1992, 141, 1349–1360. [Google Scholar] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Annu. Rev. Cell. Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; McNeil, P.L. Mechanical injury and repair of cells. Crit. Care Med. 2003, 31, S496–S501. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell. Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell. Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Blazek, A.D.; Paleo, B.J.; Weisleder, N. Plasma Membrane Repair: A Central Process for Maintaining Cellular Homeostasis. Physiology 2015, 30, 438–448. [Google Scholar] [CrossRef]
- Davenport, N.R.; Sonnemann, K.J.; Eliceiri, K.W.; Bement, W.M. Membrane dynamics during cellular wound repair. Mol. Biol. Cell 2016, 27, 2272–2285. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, X.; Yu, L.; Xu, H. Calcium signaling in membrane repair. Semin. Cell. Dev. Biol. 2015, 45, 24–31. [Google Scholar] [CrossRef]
- Nakamura, M.; Dominguez, A.N.M.; Decker, J.R.; Hull, A.J.; Verboon, J.M.; Parkhurst, S.M. Into the breach: How cells cope with wounds. Open Biol. 2018, 8, 180135. [Google Scholar] [CrossRef]
- Draeger, A.; Schoenauer, R.; Atanassoff, A.P.; Wolfmeier, H.; Babiychuk, E.B. Dealing with damage: Plasma membrane repair mechanisms. Biochimie 2014, 107, 66–72. [Google Scholar] [CrossRef]
- Lek, A.; Evesson, F.J.; Lemckert, F.A.; Redpath, G.M.; Lueders, A.K.; Turnbull, L.; Whitchurch, C.B.; North, K.N.; Cooper, S.T. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 2013, 33, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.; Ko, J.K.; Weisleder, N.; Takeshima, H.; Ma, J. Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am. J. Physiol. Physiol. 2011, 301, C106–C114. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.R.; Campbell, K.P.; Glass, D.J. MG53’s new identity. Skelet. Muscle 2013, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.-K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nature 2009, 11, 56–64. [Google Scholar]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef]
- Rezvanpour, A.; Santamaria-Kisiel, L.; Shaw, G.S. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J. Biol. Chem. 2011, 286, 40174–40183. [Google Scholar] [CrossRef]
- Inada, R.; Matsuki, M.; Yamada, K.; Morishima, Y.; Shen, S.C.; Kuramoto, N.; Yasuno, H.; Takahashi, K.; Miyachi, Y.; Yamanishi, K. Facilitated wound healing by activation of the Transglutaminase 1 gene. Am. J. Pathol. 2000, 157, 1875–1882. [Google Scholar] [CrossRef]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell. Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Kakrabarti, S.; Kobayashi, K.S.; Flavell, R.A.; Marks, C.B.; Miyake, K.; Liston, D.R.; Fowler, K.T.; Gorelick, F.S.; Andrews, N.W. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J. Cell Biol. 2003, 162, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, X.; Gao, Q.; Samie, M.A.; Azar, M.; Tsang, W.L.; Dong, L.; Sahoo, N.; Li, X.; Zhuo, Y.; et al. The intracellular Ca2+ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 2014, 20, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Greune, L.; Kahms, M.; Mildner, K.; Franzkoch, R.; Psathaki, O.E.; Zobel, T.; Zeuschner, D.; Klingauf, J.; Gerke, V. Early Endosomes Act as Local Exocytosis Hubs to Repair Endothelial Membrane Damage. Adv. Sci. 2023, 10, e2300244. [Google Scholar] [CrossRef]
- Manneville, J.-B.; Etienne-Manneville, S.; Skehel, P.; Carter, T.; Ogden, D.; Ferenczi, M. Interaction of the actin cytoskeleton with microtubules regulates secretory organelle movement near the plasma membrane in human endothelial cells. J. Cell Sci. 2003, 116, 3927–3938. [Google Scholar] [CrossRef]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Ammendolia, D.A.; Bement, W.M.; Brumell, J.H. Plasma membrane integrity: Implications for health and disease. BMC Biol. 2021, 19, 71. [Google Scholar] [CrossRef]
- Li, X.; Xiao, Y.; Cui, Y.; Tan, T.; Narasimhulu, C.A.; Hao, H.; Liu, L.; Zhang, J.; He, G.; Verfaillie, C.M.; et al. Cell membrane damage is involved in the impaired survival of bone marrow stem cells by oxidized low-density lipoprotein. J. Cell. Mol. Med. 2014, 18, 2445–2453. [Google Scholar] [CrossRef]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 5155. [Google Scholar] [CrossRef]
- Flores-Díaz, M.; Monturiol-Gross, L.; Naylor, C.; Alape-Girón, A.; Flieger, A. Bacterial sphingomyelinases and phospholipases as virulence factors. Microbiol. Mol. Biol. Rev. 2016, 80, 597–628. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Höfs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L. Membrane leakiness after viral infection and a new approach to the development of antiviral agents. Nature 1978, 272, 694–699. [Google Scholar] [CrossRef]
- Dudkina, N.V.; Spicer, B.A.; Reboul, C.F.; Conroy, P.J.; Lukoyanova, N.; Elmlund, H.; Law, R.H.P.; Ekkel, S.M.; Kondos, S.C.; Goode, R.J.A.; et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat. Commun. 2016, 7, 10588. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Bultynck, G.; Savvides, S.N. Pore-forming proteins as drivers of membrane permeabilization in cell death pathways. Nat. Rev. Mol. Cell Biol. 2023, 24, 312–333. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Bernatchez, P.N.; Acevedo, L.; Fernandez-Hernando, C.; Murata, T.; Chalouni, C.; Kim, J.; Erdjument-Bromage, H.; Shah, V.; Gratton, J.-P.; McNally, E.M.; et al. Myoferlin Regulates Vascular Endothelial Growth Factor Receptor-2 Stability and Function. J. Biol. Chem. 2007, 282, 30745–30753. [Google Scholar] [CrossRef]
- Tekin, M.; Akcayoz, D.; Incesulu, A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am. J. Med. Genet. Part A 2005, 138, 6–10. [Google Scholar] [CrossRef]
- Li, Y.; Massey, K.; Witkiewicz, H.; Schnitzer, E.J. Systems analysis of endothelial cell plasma membrane proteome of rat lung microvasculature. Proteome Sci. 2011, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Han, W.-Q.; Xia, M.; Xu, M.; Boini, K.M.; Ritter, J.K.; Li, N.-J.; Li, P.-L. Lysosome fusion to the cell membrane is mediated by the dysferlin C2A domain in coronary arterial endothelial cells. J. Cell Sci. 2012, 125, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, P.N.; Sharma, A.; Kodaman, P.; Sessa, W.C. Myoferlin is critical for endocytosis in endothelial cells. Am. J. Physiol. Physiol. 2009, 297, C484–C492. [Google Scholar] [CrossRef] [PubMed]
- Häger, S.C.; Nylandsted, J. Annexins: Players of single cell wound healing and regeneration. Commun. Integr. Biol. 2019, 12, 162–165. [Google Scholar] [CrossRef]
- Koerdt, S.N.; Ashraf, A.P.K.; Gerke, V. Annexins and plasma membrane repair. Curr. Top. Membr. 2019, 84, 43–65. [Google Scholar]
- Koerdt, S.N.; Gerke, V. Annexin A2 is involved in Ca 2+-dependent plasma membrane repair in primary human endothelial cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1046–1053. [Google Scholar] [CrossRef]
- Ashraf, A.P.K.; Gerke, V. Plasma membrane wound repair is characterized by extensive membrane lipid and protein rearrangements in vascular endothelial cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 118991. [Google Scholar] [CrossRef]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef]
- Goligorsky, M.S.; Patschan, D.; Kuo, M.C. Weibel-Palade bodies—Sentinels of acute stress. Nat. Rev. Nephrol. 2009, 5, 423–426. [Google Scholar] [CrossRef]
- Riedl Khursigara, M.; Schlam, D.; Noone, D.G.; Bruno, V.; Ortiz-Sandoval, C.G.; Pluthero, F.G.; Kahr, W.H.A.; Bowman, M.L.; James, P.; Grinstein, S.; et al. Vascular endothelial cells evade complement-mediated membrane injury via Weibel-Palade body mobilization. J. Thromb. Haemost. 2020, 18, 1484–1494. [Google Scholar] [CrossRef]
- Chehab, T.; Santos, N.C.; Holthenrich, A.; Koerdt, S.N.; Disse, J.; Schuberth, C.; Nazmi, A.R.; Neeft, M.; Koch, H.; Man, K.N.M.; et al. A novel Munc13-4/S100A10/annexin A2 complex promotes Weibel-Palade body exocytosis in endothelial cells. Mol. Biol. Cell. 2017, 28, 1688–1700. [Google Scholar] [CrossRef]
- Holthenrich, A.; Gerke, V. Regulation of von-Willebrand Factor Secretion from Endothelial Cells by the Annexin A2-S100A10 Complex. Int. J. Mol. Sci. 2018, 19, 1752. [Google Scholar] [CrossRef]
- Vieira, O.V. Rab3a and Rab10 are regulators of lysosome exocytosis and plasma membrane repair. Small GTPases 2018, 9, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Encarnação, M.; Espada, L.; Escrevente, C.; Mateus, D.; Ramalho, J.; Michelet, X.; Santarino, I.; Hsu, V.W.; Brenner, M.B.; Barral, D.C.; et al. A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J. Cell. Biol. 2016, 213, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Steinhardt, R.A. Nonmuscle Myosin IIA and IIB Have Distinct Functions in the Exocytosis-dependent Process of Cell Membrane Repair. Mol. Biol. Cell 2004, 15, 688–695. [Google Scholar] [CrossRef]
- Gregor, L.; Stock, S.; Kobold, S. ESCRT machinery: Role of membrane repair mechanisms in escaping cell death. Signal Transduct. Target. Ther. 2022, 7, 238. [Google Scholar] [CrossRef]
- Griffin, D.A.; Johnson, R.W.; Whitlock, J.M.; Pozsgai, E.R.; Heller, K.N.; Grose, W.E.; Arnold, W.D.; Sahenk, Z.; Hartzell, H.C.; Rodino-Klapac, L.R. Defective membrane fusion and repair in Anoctamin5-deficient muscular dystrophy. Hum. Mol. Genet. 2016, 25, 1900–1911. [Google Scholar] [CrossRef]
- Corrotte, M.; Cerasoli, M.; Maeda, F.Y.; Andrews, N.W. Endophilin-A2-dependent tubular endocytosis promotes plasma membrane repair and parasite invasion. J. Cell. Sci. 2020, 134, jcs249524. [Google Scholar] [CrossRef]
- White, Z.; Milad, N.; Sellers, S.L.; Bernatchez, P. Effect of Dysferlin Deficiency on Atherosclerosis and Plasma Lipoprotein Composition Under Normal and Hyperlipidemic Conditions. Front. Physiol. 2021, 12, 675322. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Yu, J.; Suárez, Y.; Rahner, C.; Dávalos, A.; Lasunción, M.A.; Sessa, W.C. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell. Metab. 2009, 10, 48–54. [Google Scholar] [CrossRef]
- Drechsler, M.; de Jong, R.; Rossaint, J.; Viola, J.R.; Leoni, G.; Wang, J.M.; Grommes, J.; Hinkel, R.; Kupatt, C.; Weber, C.; et al. Annexin A1 Counteracts Chemokine-Induced Arterial Myeloid Cell Recruitment. Circ. Res. 2015, 116, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, Q.; Han, L.; Cao, K.; Lan, T.; Xu, Z.; Wang, Y.; Gao, Y.; Xue, J.; Shan, F.; et al. Tenascin-c renders a proangiogenic phenotype in macrophage via annexin II. J. Cell. Mol. Med. 2017, 22, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Hedhli, N.; Falcone, D.J.; Huang, B.; Cesarman-Maus, G.; Kraemer, R.; Zhai, H.; Tsirka, S.E.; Santambrogio, L.; Hajjar, K.A. The annexin A2/S100A10 system in health and disease: Emerging paradigms. J. Biomed. Biotechnol. 2012, 2012, 406273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, G.; Bhat, O.M.; Li, X.; Zhang, Y.; Li, P.L. Impairment of Ceramide-Mediated Endothelial Instant Membrane Resealing During Diabetes Mellitus. Front. Physiol. 2022, 13, 910339. [Google Scholar] [CrossRef]
- Howard, A.C.; McNeil, A.K.; Xiong, F.; Xiong, W.C.; McNeil, P.L. A novel cellular defect in diabetes: Membrane repair failure. Diabetes 2011, 60, 3034–3043. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, K.; Lin, P.; Lieber, G.; Nishi, M.; Yan, R.; Wang, Z.; Yao, Y.; Li, Y.; Whitson, B.A.; et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 2014, 5, 4387. [Google Scholar] [CrossRef]
- Nagre, N.; Cong, X.; Ji, H.-L.; Schreiber, J.M.; Fu, H.; Pepper, I.; Warren, S.; Sill, J.M.; Hubmayr, R.D.; Zhao, X. Inhaled TRIM72 Protein Protects Ventilation Injury to the Lung through Injury-guided Cell Repair. Am. J. Respir. Cell Mol. Biol. 2018, 59, 635–647. [Google Scholar] [CrossRef]
- Wang, X.; Xie, W.; Zhang, Y.; Lin, P.; Han, L.; Han, P.; Wang, Y.; Chen, Z.; Ji, G.; Zheng, M.; et al. Cardioprotection of Ischemia/Reperfusion Injury by Cholesterol-Dependent MG53-Mediated Membrane Repair. Circ. Res. 2010, 107, 76–83. [Google Scholar]
- Gushchina, L.V.; Bhattacharya, S.; McElhanon, K.E.; Choi, J.H.; Manring, H.; Beck, E.X.; Alloush, J.; Weisleder, N. Treatment with Recombinant Human MG53 Protein Increases Membrane Integrity in a Mouse Model of Limb Girdle Muscular Dystrophy 2B. Mol. Ther. 2017, 25, 2360–2371. [Google Scholar] [CrossRef]
- Guan, F.; Huang, T.; Wang, X.; Xing, Q.; Gumpper, K.; Li, P.; Song, J.; Tan, T.; Yang, G.L.; Zang, X.; et al. The TRIM protein Mitsugumin 53 enhances survival and therapeutic efficacy of stem cells in murine traumatic brain injury. Stem. Cell. Res. Ther. 2019, 10, 352. [Google Scholar] [CrossRef]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra36. [Google Scholar] [CrossRef]
- Yao, W.; Li, H.; Han, X.; Chen, C.; Zhang, Y.; Tai, W.L.; Xia, Z.; Hei, Z. MG53 anchored by dysferlin to cell membrane reduces hepatocyte apoptosis which induced by ischaemia/reperfusion injury in vivo and in vitro. J. Cell. Mol. Med. 2017, 21, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Barbero, N.; San Sebastian-Jaraba, I.; Blázquez-Serra, R.; Martín-Ventura, J.L.; Blanco-Colio, L.M. Annexins and cardiovascular diseases: Beyond membrane trafficking and repair. Front Cell. Dev. Biol. 2022, 10, 1000760. [Google Scholar] [CrossRef]
- Bao, H.J.; Wang, T.; Zhang, M.Y.; Liu, R.; Dai, D.K.; Wang, Y.Q.; Wang, L.; Zhang, L.; Gao, Y.Z.; Qin, Z.H.; et al. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem. Res. 2012, 37, 2856–2867. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, R.A.; Alderton, J.M. Poloxamer 188 Enhances Endothelial Cell Survival in Bovine Corneas in Cold Storage. Cornea 2006, 25, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Moloughney, J.G.; Weisleder, N. Poloxamer 188 (p188) as a membrane resealing reagent in biomedical applications. Recent Pat. Biotechnol. 2012, 6, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell. Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef]
- Nozaki, K.; Maltez, V.I.; Rayamajhi, M.; Tubbs, A.L.; Mitchell, J.E.; Lacey, C.A.; Harvest, C.K.; Li, L.; Nash, W.T.; Larson, H.N.; et al. Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 2022, 606, 960–967. [Google Scholar] [CrossRef]


{kind=link}
| Name | Action | Experimental Conditions | References |
|---|---|---|---|
| Reconbinant MG53 | Increased membrane integrity and protects against organ injury | Mice, in vivo injection | [67,68,69,70,71,72] |
| Recombinant annexins | Improve the outcome of sepsis, myocardial infarction, and ischemia-reperfusion injury | Mice, in vivo injection | |
| Clinical trial | [73] | ||
| Poloxamer 188 | Reduce blood viscosity for transfusion | FDA-approved | |
| Improve outcome of TBI | Mice, in vivo injection | [76] | |
| Prolong endothelial cell survival | Bovine corneas, in vitro | [77] | |
| Acid sphingomyelinase | Promote membrane resealing | Cells, in vitro | [78] |
| Sphingomyelin and ceramide | Protect endothelial cell membrane impairment during diabetes | Cells, in vitro Mice, in vivo | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zha, D.; Wang, S.; Monaghan-Nichols, P.; Qian, Y.; Sampath, V.; Fu, M. Mechanisms of Endothelial Cell Membrane Repair: Progress and Perspectives. Cells 2023, 12, 2648. https://doi.org/10.3390/cells12222648
Zha D, Wang S, Monaghan-Nichols P, Qian Y, Sampath V, Fu M. Mechanisms of Endothelial Cell Membrane Repair: Progress and Perspectives. Cells. 2023; 12(22):2648. https://doi.org/10.3390/cells12222648
Chicago/Turabian StyleZha, Duoduo, Shizhen Wang, Paula Monaghan-Nichols, Yisong Qian, Venkatesh Sampath, and Mingui Fu. 2023. "Mechanisms of Endothelial Cell Membrane Repair: Progress and Perspectives" Cells 12, no. 22: 2648. https://doi.org/10.3390/cells12222648