
Panobinostat Induced Spatial In Situ Biomarkers Predictive of Anti-PD-1 Efficacy in Mouse Mammary Carcinoma
 ,
,
Abstract
:1. Introduction
1.1. Immune Checkpoint Blockade and Its Clinical Biomarkers in Breast Cancer
1.2. Immunogenic Cell Death Sensitizes Tumors to ICB Efficacy
1.3. Panobinostat Was Predicted to Induce ICD and Synergize with Anti-PD-1 in Breast Cancer Using a Multiplex Implantable Microdevice Assay System
2. Materials and Methods
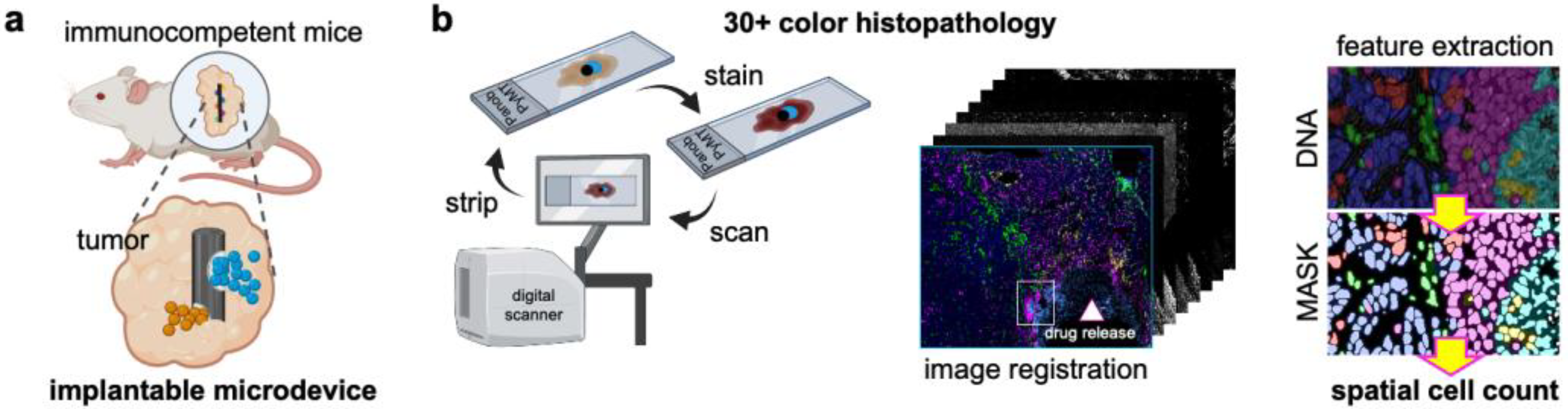
2.1. Murine Models and Studies
2.2. Microdevice Implantation and Sample Collection
2.3. Multiplex Immunohistochemistry (mIHC)
2.4. Image Processing and Feature Extraction of mIHC Images
2.5. Spatial Cell Analysis In Situ
2.6. Code and Data Availability
3. Results
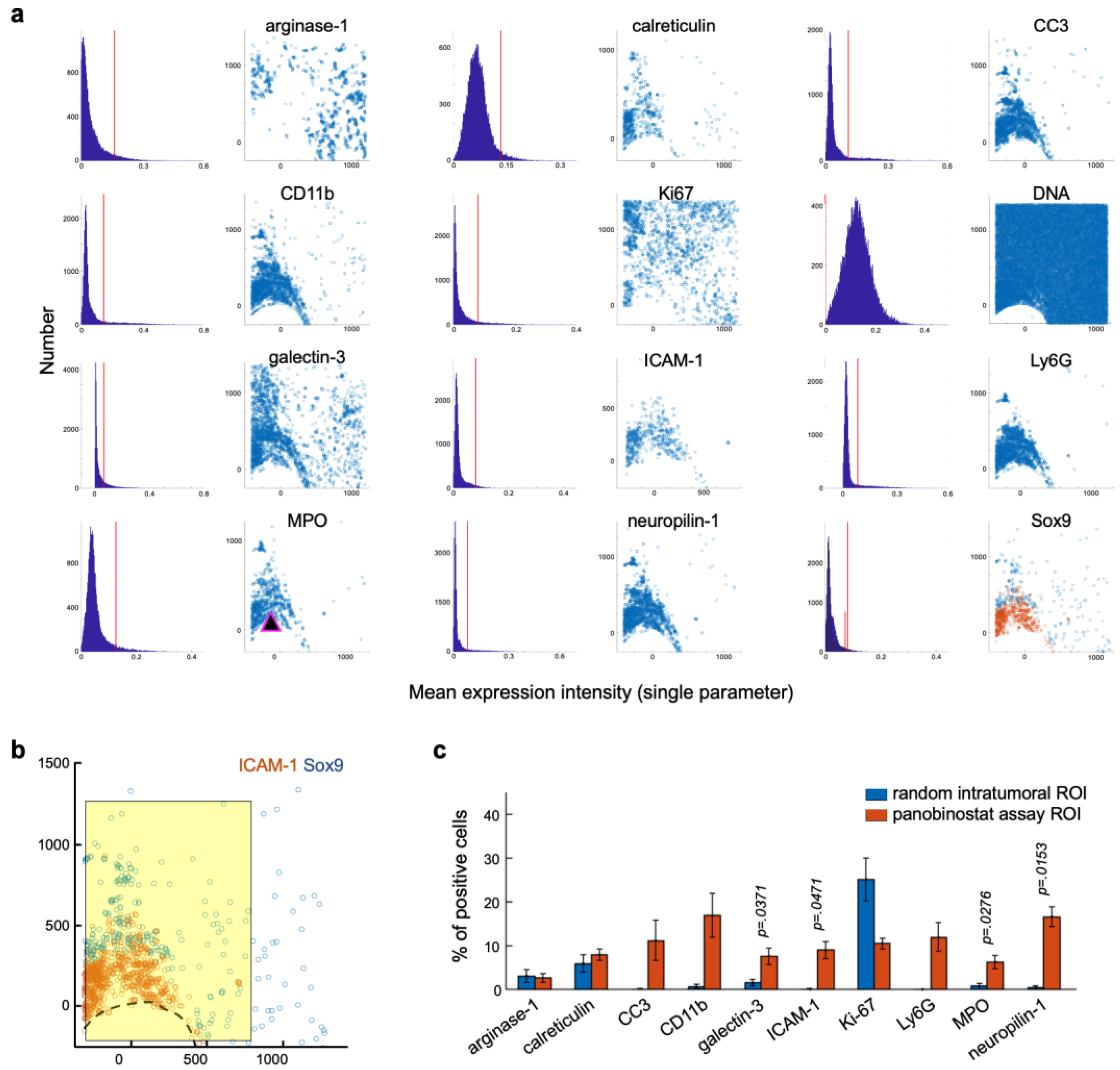
3.1. Literature Review to Find Biomarkers of Ongoing/Induced Anti-Tumor Immunity
3.2. Components of the Multiplex Implantable Microdevice Assay (MIMA)
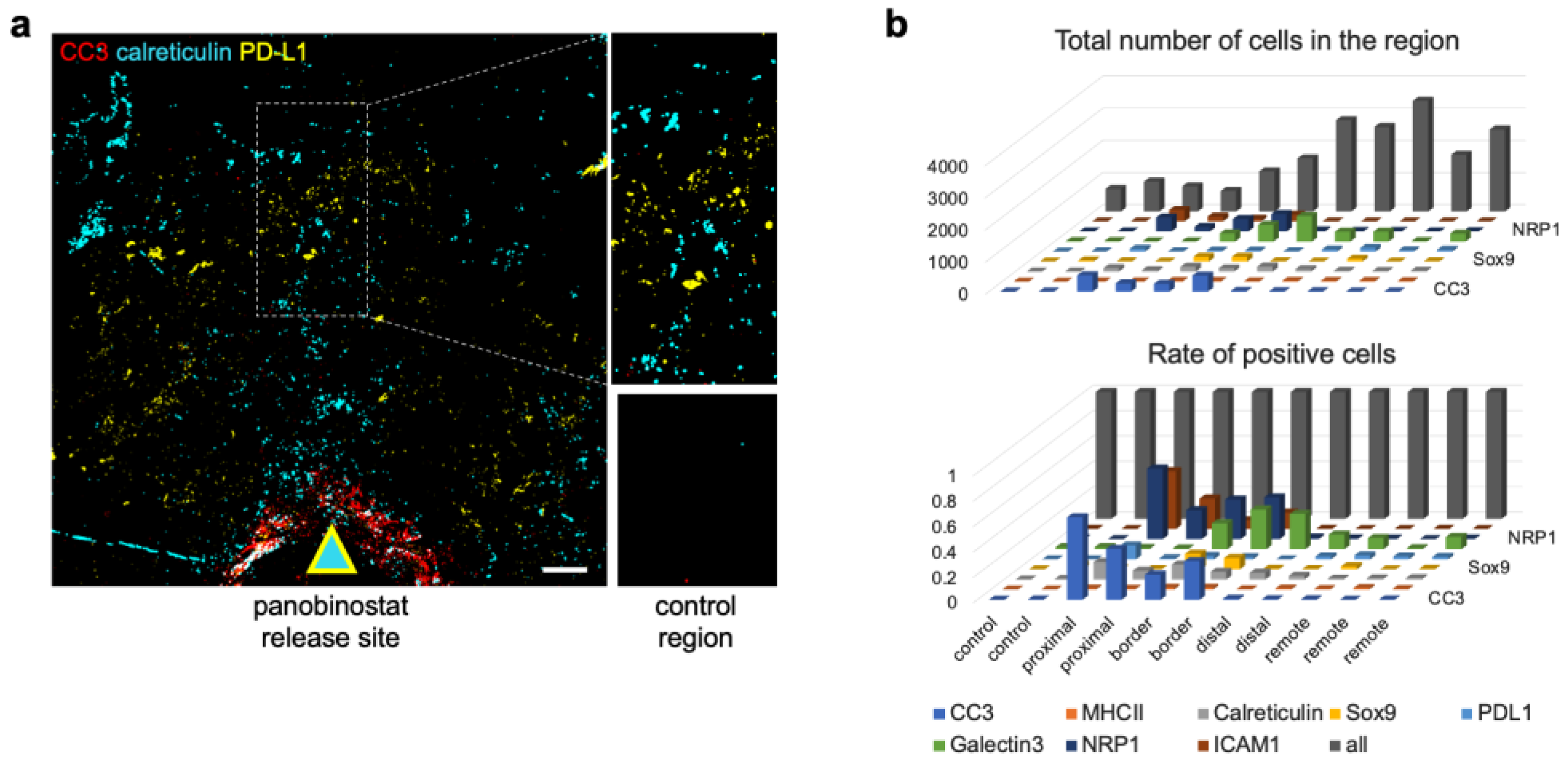
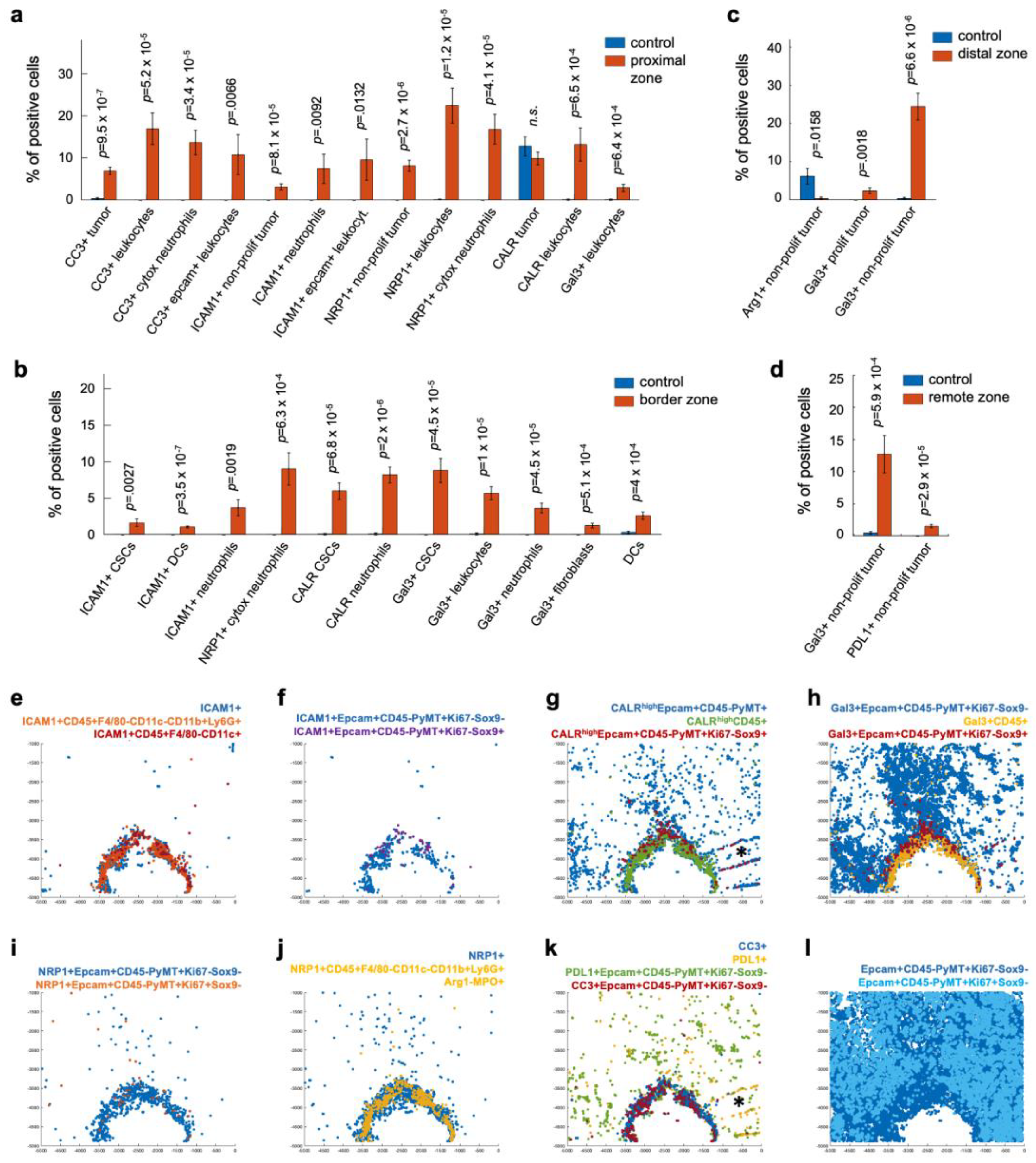
3.3. Candidate Biomarkers Were Spatially Associated with Panobinostat Drug Release Site
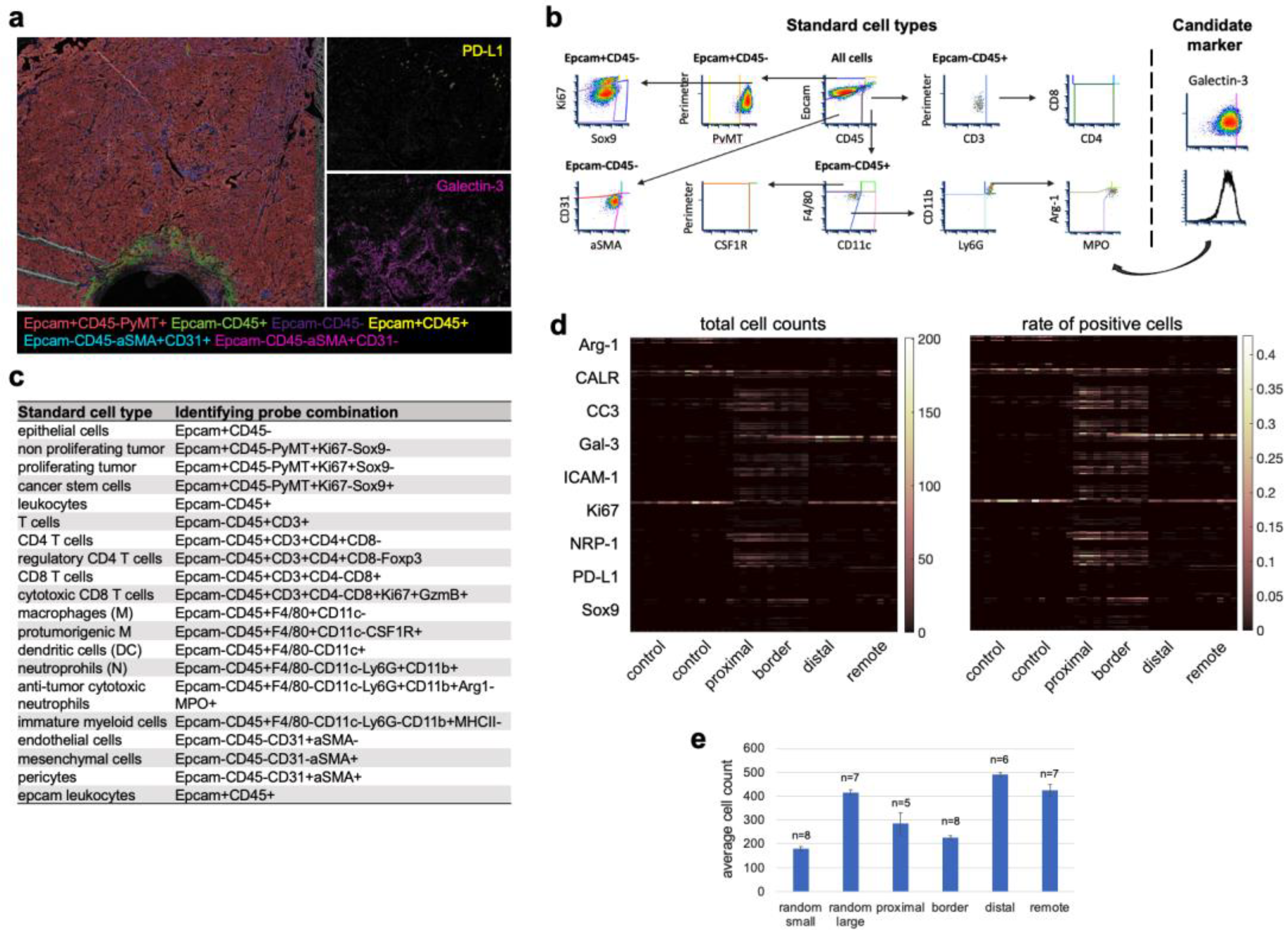
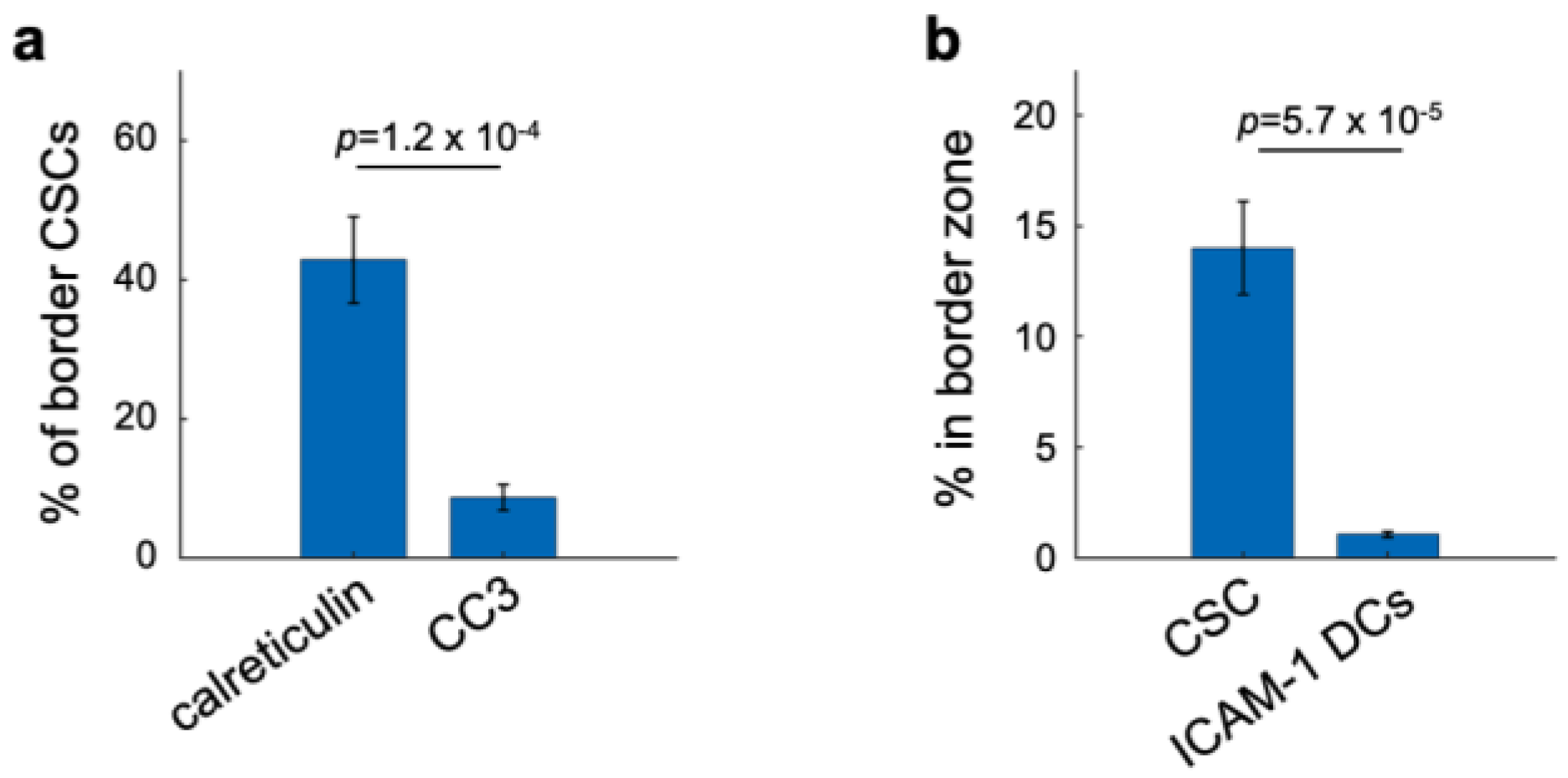
3.4. Cell Type Classification of the Candidate Biomarkers in Space Defines Their Roles during ICD
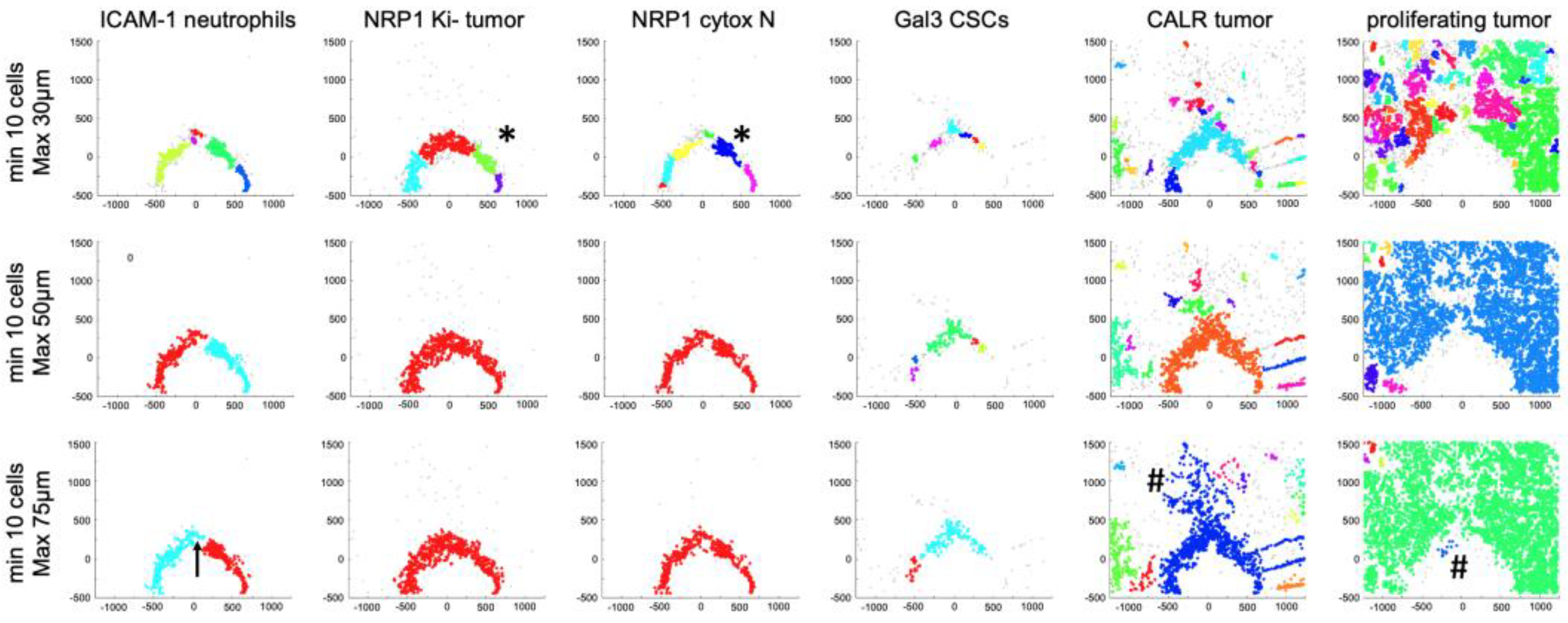
3.5. Spatial Clustering of Marker Combinations Serves as a Treatment-Specific Biomarker with Predictive Value
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tatarova, Z.; Blumberg, D.C.; Korkola, J.E.; Heiser, L.M.; Muschler, J.L.; Schedin, P.J.; Ahn, S.W.; Mills, G.B.; Coussens, L.M.; Jonas, O.; et al. A Multiplex Implantable Microdevice Assay Identifies Synergistic Combinations of Cancer Immunotherapies and Conventional Drugs. Nat. Biotechnol. 2022, 40, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Coxon, A.T.; Dunn, G.P. Therapeutic Applications of the Cancer Immunoediting Hypothesis. Semin. Cancer Biol. 2022, 78, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J. Putting the Immunologic Brakes on Cancer. Cell 2018, 175, 1452–1454. [Google Scholar] [CrossRef] [Green Version]
- McArthur, H.L.; Page, D.B. Immunotherapy for the Treatment of Breast Cancer: Checkpoint Blockade, Cancer Vaccines, and Future Directions in Combination Immunotherapy. Clin. Adv. Hematol. Oncol. 2016, 14, 922–933. [Google Scholar]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The Hallmarks of Successful Anticancer Immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef]
- El Bairi, K.; Haynes, H.R.; Blackley, E.; Fineberg, S.; Shear, J.; Turner, S.; de Freitas, J.R.; Sur, D.; Amendola, L.C.; Gharib, M.; et al. The Tale of TILs in Breast Cancer: A Report from The International Immuno-Oncology Biomarker Working Group. Nat. Partn. J. Breast Cancer 2021, 7, 150. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-Infiltrating Lymphocytes and Prognosis in Different Subtypes of Breast Cancer: A Pooled Analysis of 3771 Patients Treated with Neoadjuvant Therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Curigliano, G. Recent Advances in Triple Negative Breast Cancer: The Immunotherapy Era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Ou-Yang, F.; Li, C.-L.; Chen, C.-C.; Shen, Y.-C.; Moi, S.-H.; Luo, C.-W.; Xia, W.-Y.; Wang, Y.-N.; Lee, H.-H.; Wang, L.-H.; et al. De-Glycosylated Membrane PD-L1 in Tumor Tissues as a Biomarker for Responsiveness to Atezolizumab (Tecentriq) in Advanced Breast Cancer Patients. Am. J. Cancer Res. 2022, 12, 123–137. [Google Scholar] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Md Sakib Hossain, D.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib Induces Immunogenic Cell Death and Enhances Anti- PD1–Mediated Tumor Suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming Anti-Tumor Immunity by Radiotherapy: Dying Tumor Cell-Derived DAMPs Trigger Endothelial Cell Activation and Recruitment of Myeloid Cells. Oncoimmunology 2019, 8, e1523097. [Google Scholar] [CrossRef] [Green Version]
- Van Vloten, J.P.; Workenhe, S.T.; Wootton, S.K.; Mossman, K.L.; Bridle, B.W. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J. Immunol. 2018, 200, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Dong, W.; Li, M.; Guo, Z.; Wang, Q.; Liu, Y.; Bi, Y.; Zhou, H.; Wang, Y. Nitric Oxide Induces Immunogenic Cell Death and Potentiates Cancer Immunotherapy. ACS Nano 2022, 16, 3881–3894. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus Guidelines for the Detection of Immunogenic Cell Death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Denardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Sharfaa, A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic Cell Stress and Death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Rufo, N.; Korovesis, D.; Van Eygen, S.; Derua, R.; Garg, A.D.; Finotello, F.; Vara-Perez, M.; Rožanc, J.; Dewaele, M.; de Witte, P.A.; et al. Stress-Induced Inflammation Evoked by Immunogenic Cell Death Is Blunted by the IRE1α Kinase Inhibitor KIRA6 through HSP60 Targeting. Cell Death Differ. 2022, 29, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Fang, S.; Fasche, T.; Van Eygen, S.; Maes, J.; Van Woensel, M.; Koks, C.; Vanthillo, N.; Graf, N.; et al. Pathogen Response-like Recruitment and Activation of Neutrophils by Sterile Immunogenic Dying Cells Drives Neutrophil-Mediated Residual Cell Killing. Cell Death Differ. 2017, 24, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunogenic Cell Death Induction by Anticancer Chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oltean, T.; Lippens, L.; Lemeire, K.; De Tender, C. Association of Cell Death Markers with Tumor Immune Cell Infiltrates After Chemo-Radiation in Cervical Cancer. Front. Oncol. 2022, 12, 892813. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Tsujikawa, T.; Margolin, A.; Coussens, L.M.; Gray, J.W. Multiplexed Immunohistochemistry Image Analysis Using Sparse Coding. In Proceedings of the 2017 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Jeju, Korea, 11–15 July 2017; pp. 4046–4049. [Google Scholar] [CrossRef]
- Eng, J.; Thibault, G.; Luoh, S.-W.; Gray, J.W.; Chang, Y.H.; Chin, K. Cyclic Multiplexed-Immunofluorescence (CmIF), a Highly Multiplexed Method for Single-Cell Analysis. In Biomarkers for Immunotherapy of Cancer: Methods and Protocols; Thurin, M., Cesano, A., Marincola, F.M., Eds.; Springer: New York, NY, USA, 2020; pp. 521–562. [Google Scholar] [CrossRef]
- Li, A.; Keck, J.M.; Parmar, S.; Patterson, J.; Labrie, M.; Creason, A.L.; Johnson, B.E.; Downey, M.; Thomas, G.; Beadling, C.; et al. Characterizing Advanced Breast Cancer Heterogeneity and Treatment Resistance through Serial Biopsies and Comprehensive Analytics. NPJ Precis. Oncol. 2021, 5, 28. [Google Scholar] [CrossRef]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The Single-Cell Pathology Landscape of Breast Cancer. Nature 2020, 578, 615–620. [Google Scholar] [CrossRef]
- Jonas, O.; Landry, H.M.; Fuller, J.E.; Santini, J.T.; Baselga, J.; Tepper, R.I.; Cima, M.J.; Langer, R. An Implantable Microdevice to Perform High-Throughput in Vivo Drug Sensitivity Testing in Tumors. Sci. Transl. Med. 2015, 7, 284ra57. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Pham, N.A.; Morrison, A.; Schwock, J.; Aviel-Ronen, S.; Lakovlev, V.; Tsao, M.S.; Ho, J.; Hedley, D.W. Quantitative Image Analysis of Immunohistochemical Stains Using a CMYK Color Model. Diagn. Pathol. 2007, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, G.; Betts, C.B.; Liudahl, S.M.; Sivagnanam, S.; Cotechini, T.; Larson, W.; Goecks, J.; Pai, S.I.; Daniel, R.; Tsujikawa, T.; et al. Chapter One—High-dimensional multiplexed immunohistochemical characterization of immune contexture in human cancers. Methods Enzymol. 2020, 635, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image Analysis Software for Identifying and Quantifying Cell Phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wan, B.; Chen, X.; Zhan, P.; Zhao, Y.; Zhang, T.; Liu, H.; Afzal, M.Z.; Dermime, S.; Hochwald, S.N.; et al. The Association of PD-L1 Expression with the Efficacy of Anti-PD-1/PD-L1 Immunotherapy and Survival of Non-Small Cell Lung Cancer Patients: A Meta-Analysis of Randomized Controlled Trials. Transl. Lung Cancer Res. 2019, 8, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Patnaik, A.; Swanson, K.D.; Csizmadia, E.; Solanki, A.; Landon-Brace, N.; Gehring, M.P.; Helenius, K.; Olson, B.M.; Pyzer, A.R.; Wang, L.C.; et al. Cabozantinib Eradicates Advanced Murine Prostate Cancer by Activating Antitumor Innate Immunity. Cancer Discov. 2017, 7, 750–765. [Google Scholar] [CrossRef] [Green Version]
- Passlick, B.; Pantel, K.; Kubuschok, B.; Angstwurm, M.; Neher, A.; Thetter, O.; Schweiberer, L.; Izbicki, J.R. Expression of MHC Molecules and ICAM-1 on Non-Small Cell Lung Carcinomas: Association with Early Lymphatic Spread of Tumour Cells. Eur. J. Cancer 1996, 32, 141–145. [Google Scholar] [CrossRef]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-Dimensional Single-Cell Analysis Predicts Response to Anti-PD-1 Immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef]
- Lanzavecchla, A. Licence to Kill. Nature 1998, 393, 413–414. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-Associated Neutrophils Display a Distinct N1 Profile Following TGFβ Modulation: A Transcriptomics Analysis of pro- vs. Antitumor TANs. Oncoimmunology 2016, 5, e1232221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the Immune System: Ins and Outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC Inhibition Reduces Breast Tumours and Metastases through Anti-Tumour Macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-Direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, M.; Martins, I.; Mann, D.M.; Finkbeiner, S.; Devys, D.; Greenberg, M.E.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 495–499. [Google Scholar] [CrossRef]
- Sato, S.; Nieminen, J. Seeing Strangers or Announcing “Danger”: Galectin-3 in Two Models of Innate Immunity. Glycoconj. J. 2002, 19, 583–591. [Google Scholar] [CrossRef]
- Karlsson, B.A.; Follin, P.; Leffler, H.; Dahlgren, C. Galectin-3 Activates the NADPH-Oxidase in Exudated but Not Peripheral Blood Neutrophils. Blood 2018, 91, 3430–3438. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Froio, R.M.; Sciuto, T.E.; Dvorak, A.M.; Alon, R.; Luscinskas, F.W. ICAM-1 Regulates Neutrophil Adhesion and Transcellular Migration of TNF-α-Activated Vascular Endothelium under Flow. Blood 2005, 106, 584–592. [Google Scholar] [CrossRef]
- Gittens, B.R.; Bodkin, J.V.; Nourshargh, S.; Perretti, M.; Cooper, D. Galectin-3: A Positive Regulator of Leukocyte Recruitment in the Inflamed Microcirculation. J. Immunol. 2018, 198, 4458–4469. [Google Scholar] [CrossRef] [Green Version]
- Kerros, C.; Tripathi, S.C.; Zha, D.; Mehrens, J.M.; Sergeeva, A.; Philips, A.V.; Qiao, N.; Peters, H.L.; Katayama, H.; Sukhumalchandra, P.; et al. Neuropilin-1 Mediates Neutrophil Elastase Uptake and Cross-Presentation in Breast Cancer Cells. J. Biol. Chem. 2017, 292, 10295–10305. [Google Scholar] [CrossRef] [Green Version]
- Tordjman, R.; Lepelletier, Y.; Lemarchandel, V.; Cambot, M.; Gaulard, P.; Hermine, O.; Roméo, P.H. A Neuronal Receptor, Neuropilin-I, Is Essential for the Initiation of the Primary Immune Response. Nat. Immunol. 2002, 3, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Attalla, S.; Taifour, T.; Bui, T.; Muller, W. Insights from Transgenic Mouse Models of PyMT-Induced Breast Cancer: Recapitulating Human Breast Cancer Progression in Vivo. Oncogene 2021, 40, 475–491. [Google Scholar] [CrossRef]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to Malignancy in the Polyoma Middle T Oncoprotein Mouse Breast Cancer Model Provides a Reliable Model for Human Diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of Mammary Tumors by Expression of Polyomavirus Middle T Oncogene: A Transgenic Mouse Model for Metastatic Disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef] [Green Version]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of Conserved Gene Expression Features between Murine Mammary Carcinoma Models and Human Breast Tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 Cooperatively Determine the Mammary Stem Cell State. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The Sterile Inflammatory Response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Sholl, L.M. Biomarkers of Response to Checkpoint Inhibitors beyond PD-L1 in Lung Cancer. Mod. Pathol. 2022, 35, 66–74. [Google Scholar] [CrossRef]
- Wendel Naumann, R.; Hollebecque, A.; Meyer, T.; Devlin, M.J.; Oaknin, A.; Kerger, J.; López-Picazo, J.M.; Machiels, J.P.; Delord, J.P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results from the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef]
- Grossman, J.E.; Vasudevan, D.; Joyce, C.E.; Hildago, M. Is PD-L1 a Consistent Biomarker for Anti-PD-1 Therapy? The Model of Balstilimab in a Virally-Driven Tumor. Oncogene 2021, 40, 1393–1395. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bifulco, C.; Bindea, G.; Marliot, F.; Lugli, A.; Lee, J.J.; Zlobec, I.; Rau, T.T.; Berger, M.D.; Nagtegaal, I.D.; et al. Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. J. Clin. Oncol. 2020, 38, 3638–3651. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-γ Drives TregFragility to Promote Anti-Tumor Immunity. Cell 2017, 169, 1130–1141.e11. [Google Scholar] [CrossRef] [Green Version]
- Matkar, P.N.; Singh, K.K.; Rudenko, D.; Kim, Y.J.; Kuliszewski, M.A.; Prud’homme, G.J.; Hedley, D.W.; Leong-Poi, H. Novel Regulatory Role of Neuropilin-1 in Endothelial-to-Mesenchymal Transition and Fibrosis in Pancreatic Ductal Adenocarcinoma. Oncotarget 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Chen, J.Y.; Weissman-Tsukamoto, R.; Volkmer, J.P.; Ho, P.Y.; McKenna, K.M.; Cheshier, S.; Zhang, M.; Guo, N.; Gip, P.; et al. Macrophages Eat Cancer Cells Using Their Own Calreticulin as a Guide: Roles of TLR and Btk. Proc. Natl. Acad. Sci. USA 2015, 112, 2145–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalbano, R.; Waldegger, P.; Quint, K.; Jabari, S.; Neureiter, D.; Illig, R.; Ocker, M.; Di Fazio, P. Endoplasmic Reticulum Stress Plays a Pivotal Role in Cell Death Mediated by the Pan-Deacetylase Inhibitor Panobinostat in Human Hepatocellular Cancer Cells. Transl. Oncol. 2013, 6, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Full/Alternative Name and Literature | Reference [37] |
|---|---|---|
| ICAM-1 | Intracellular adhesion molecule 1 | P13597 |
| Increased expression in the vascular and perivascular spaces during ICD induced by ionizing radiotherapy in BC [15] or by cabozantinib in prostate cancer [38], respectively. Absent tumor expression is prognostic of lymphatic spread in early lung cancer [39]. Melanoma patients responding to anti-PD-1 immunotherapy have higher ICAM-1 on circulating monocytes [40]. ICAM-1 is a biomarker of licensed myeloid cells [41,42] and anti-tumor neutrophils [43,44]. | Krombach et al., 2019; Patnaik et al., 2017; Passlick et al., 1996; Krieg et al., 2018; Vonderheide, 2018; Lanzavecchla, 1998; Fridlender et al., 2009; Shaul et al., 2016 | |
| CALR | calreticulin | P14211 |
| Pre-apoptotic translocation of calreticulin to the cell surface is an indicator of induced immunogenic cell death and serves as an “eat me” signal for phagocytosis by dendritic cells [21] and neutrophils [24]. Calreticulin also facilitates the folding of class I major histocompatibility complex (MHC-I) molecules, thereby affecting antigen-presenting machinery and T-cell responses [45]. | Obeid et al., 2007; Garg et al., 2017; Raghavan et al., 2013 | |
| Gal-3 | galectin-3, also known as Mac-2 | P16110 |
| A class II HDAC inhibitor shown to be synergistic with anti-PD-1-immunotherapy-induced galectin-3 expression in mammary carcinoma, as measured by IHC [46]. Galectin-3 targets and activates autophagy [47], which is required for the immunogenicity of cell death [48]. Galectin-3 is an inducible “danger signal” molecule for innate immunity [49] and activates extravasated but not peripheral blood neutrophils [50]. Exogenous galectin-3 and ICAM-1 were shown to be involved in the slow rolling of leukocytes [51] and their recruitment into the tissue from the inflamed microcirculation [52]. | Guerriero et al., 2017; Chauhan et al., 2016; Michaud et al., 2011; Sato and Nieminen, 2002; Karlsson et al., 2018; Yang et al., 2005; Gittens et al., 2018 | |
| MPO | myeloperoxidase | P11247 |
| A profound anti-tumor response to cabozantinib was accompanied by cytotoxic (MPO) neutrophil (Ly6G) infiltration in prostate cancer. In this study, cabozantinib induced tumor cell death in vivo, and ex vivo the therapy induced HMGB-1 release to the supernatant, as measured using ELISA, which was suggestive of ICD induction [38]. | Patnaik et al., 2017 | |
| NRP-1 | neuropilin-1 | P97333 |
| In breast cancer cells, neuropilin-1 is involved in cross-presentation and is essential for the killing of neutrophil elastase peptide specific cytotoxic T-cell [53]. Early studies on neuropilin-1 focused on parallels of the immune and nervous systems and showed that the protein can be expressed on DCs and resting T cells. Neuropilin-1 mediated clustering between these two populations, the polarization of the protein upon contact, and proliferation of T cells, suggesting that it is essential for the initiation of the primary immune response [54]. | Kerros et al., 2017; Tordjman et al., 2002 | |
| CC3 | Cleaved caspase-3 | NA |
| Well-established biomarker of cell death | multiple | |
| PD-L1 | Program (cell) death ligand 1, B7 homolog 1 (B7-H1) | Q9EP73 |
| Established biomarker of ICB efficacy | multiple |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatarova, Z.; Blumberg, D.C.; Bensen, A.; Mills, G.B.; Jonas, O. Panobinostat Induced Spatial In Situ Biomarkers Predictive of Anti-PD-1 Efficacy in Mouse Mammary Carcinoma. Cells 2023, 12, 308. https://doi.org/10.3390/cells12020308
Tatarova Z, Blumberg DC, Bensen A, Mills GB, Jonas O. Panobinostat Induced Spatial In Situ Biomarkers Predictive of Anti-PD-1 Efficacy in Mouse Mammary Carcinoma. Cells. 2023; 12(2):308. https://doi.org/10.3390/cells12020308
Chicago/Turabian StyleTatarova, Zuzana, Dylan C. Blumberg, AeSoon Bensen, Gordon B. Mills, and Oliver Jonas. 2023. "Panobinostat Induced Spatial In Situ Biomarkers Predictive of Anti-PD-1 Efficacy in Mouse Mammary Carcinoma" Cells 12, no. 2: 308. https://doi.org/10.3390/cells12020308