
NFκB-Mediated Expression of Phosphoinositide 3-Kinase δ Is Critical for Mesenchymal Transition in Retinal Pigment Epithelial Cells
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Major Reagents
2.2. RNA Sequencing
2.3. DNA Constructs
2.4. Lentivirus Production
2.5. Western Blotting
2.6. Immunofluorescence
2.7. Quantitative PCR
2.8. Cell Migration Assay
2.9. Luciferase Assay-PIK3CD Reporter
2.10. Chromatin Immunoprecipitation Assay
2.11. Statistics
3. Results
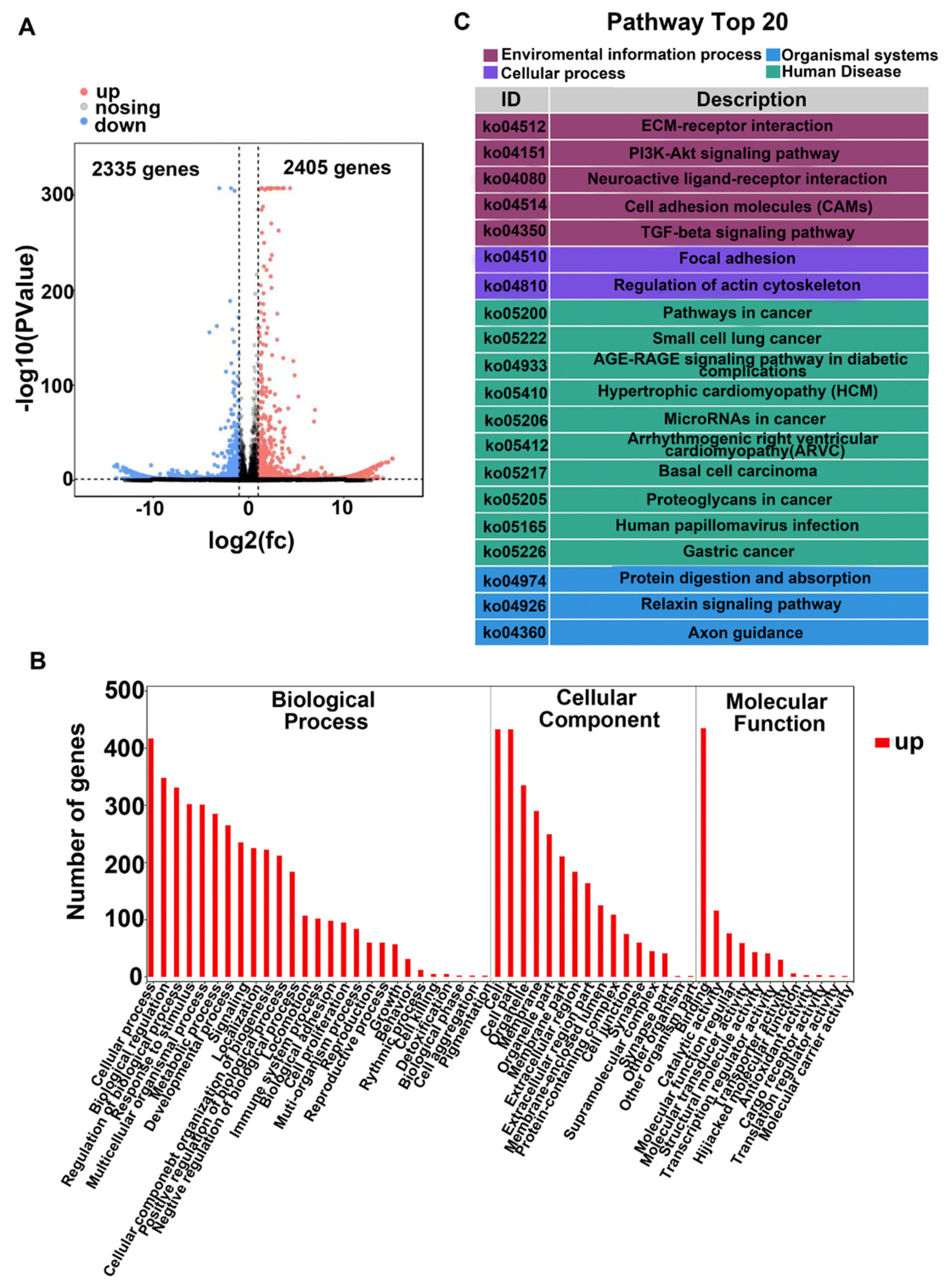
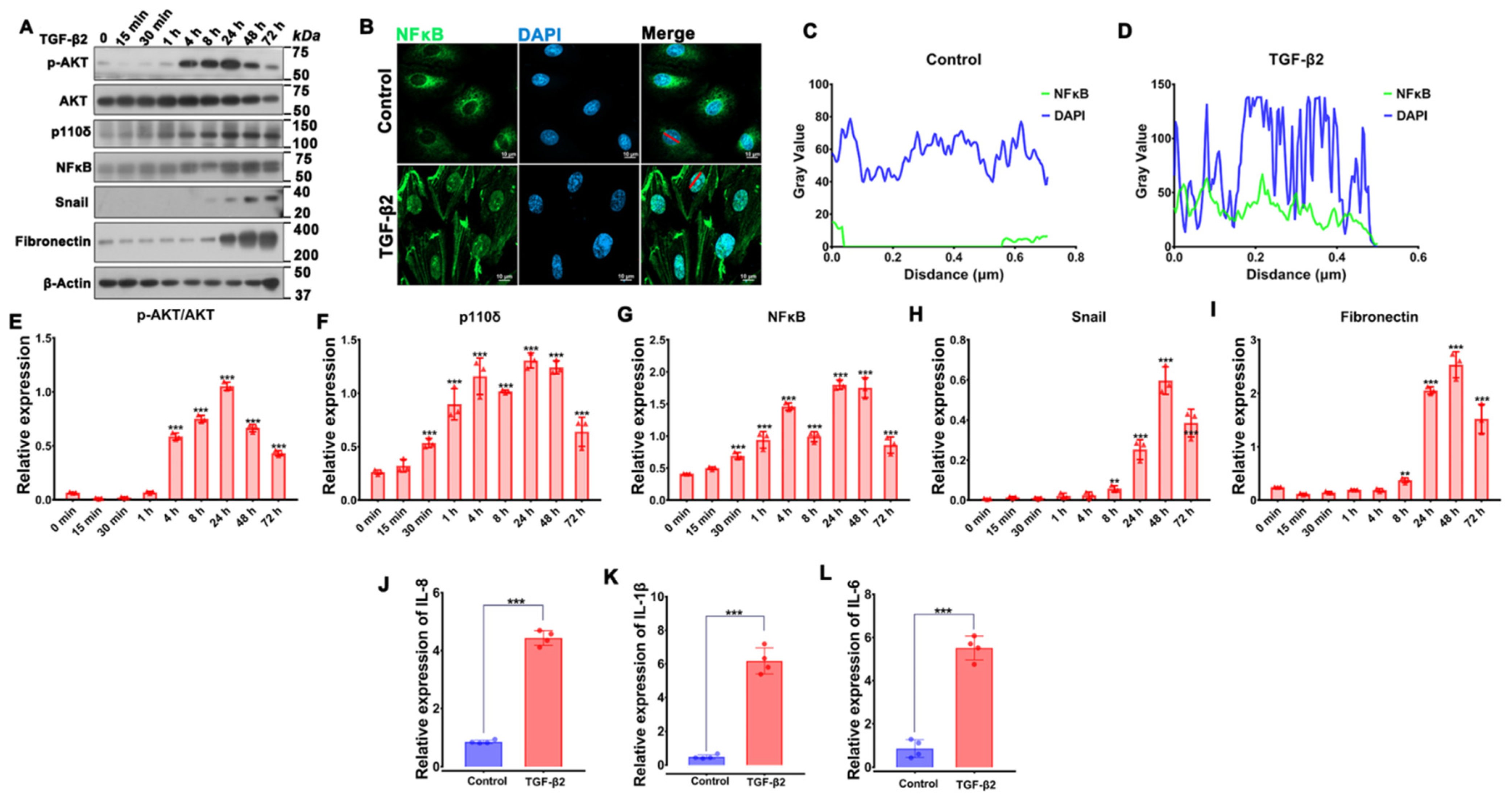
3.1. RNA Sequencing Analysis Uncovers TGF-β2-Induced PI3K/Akt Signaling in RPE Cells
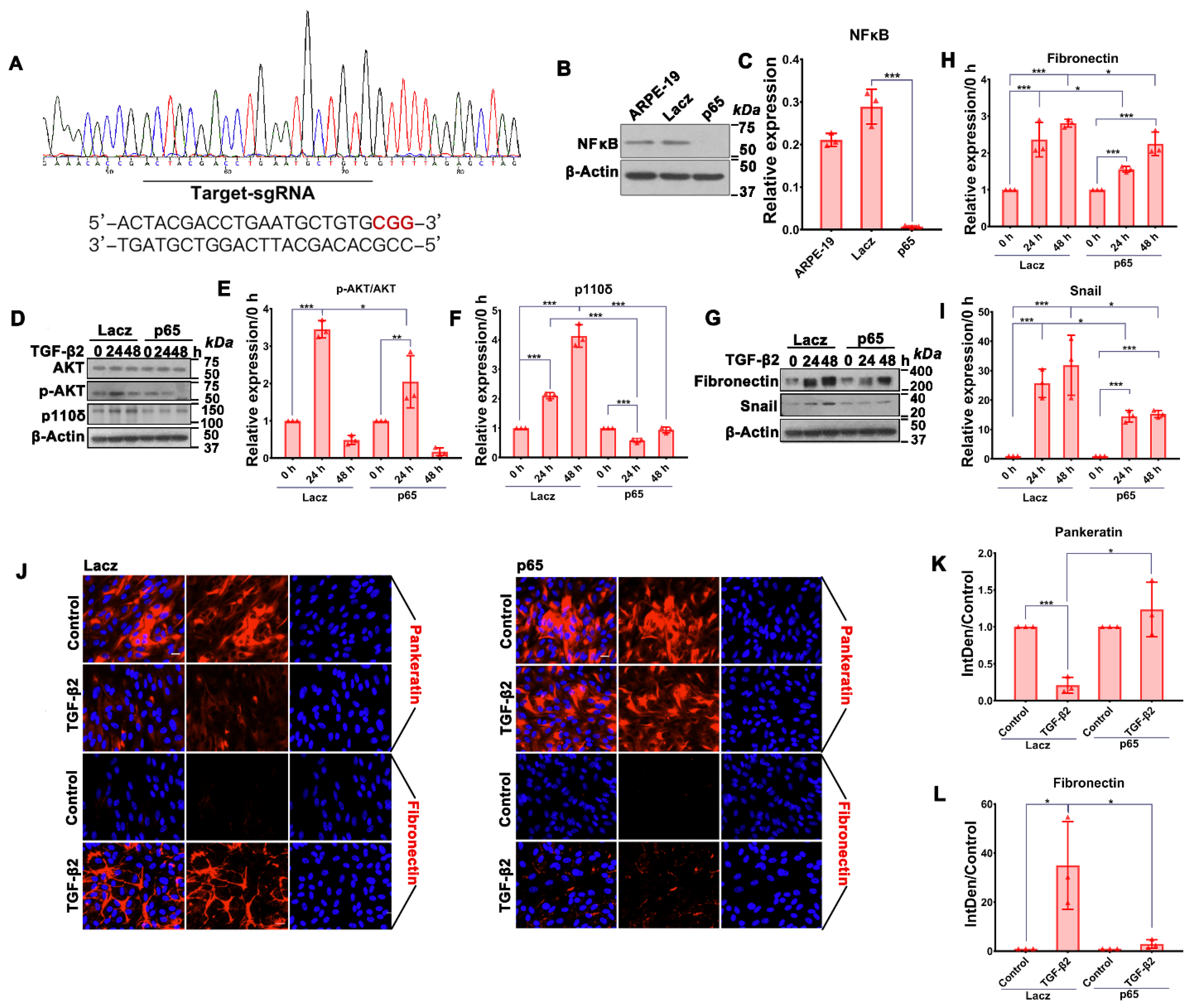
3.2. Depletion of NFκB/p65 in RPE Cells Attenuates TGF-β2-Induced Akt Activation, Expression of p110δ as Well as EMT
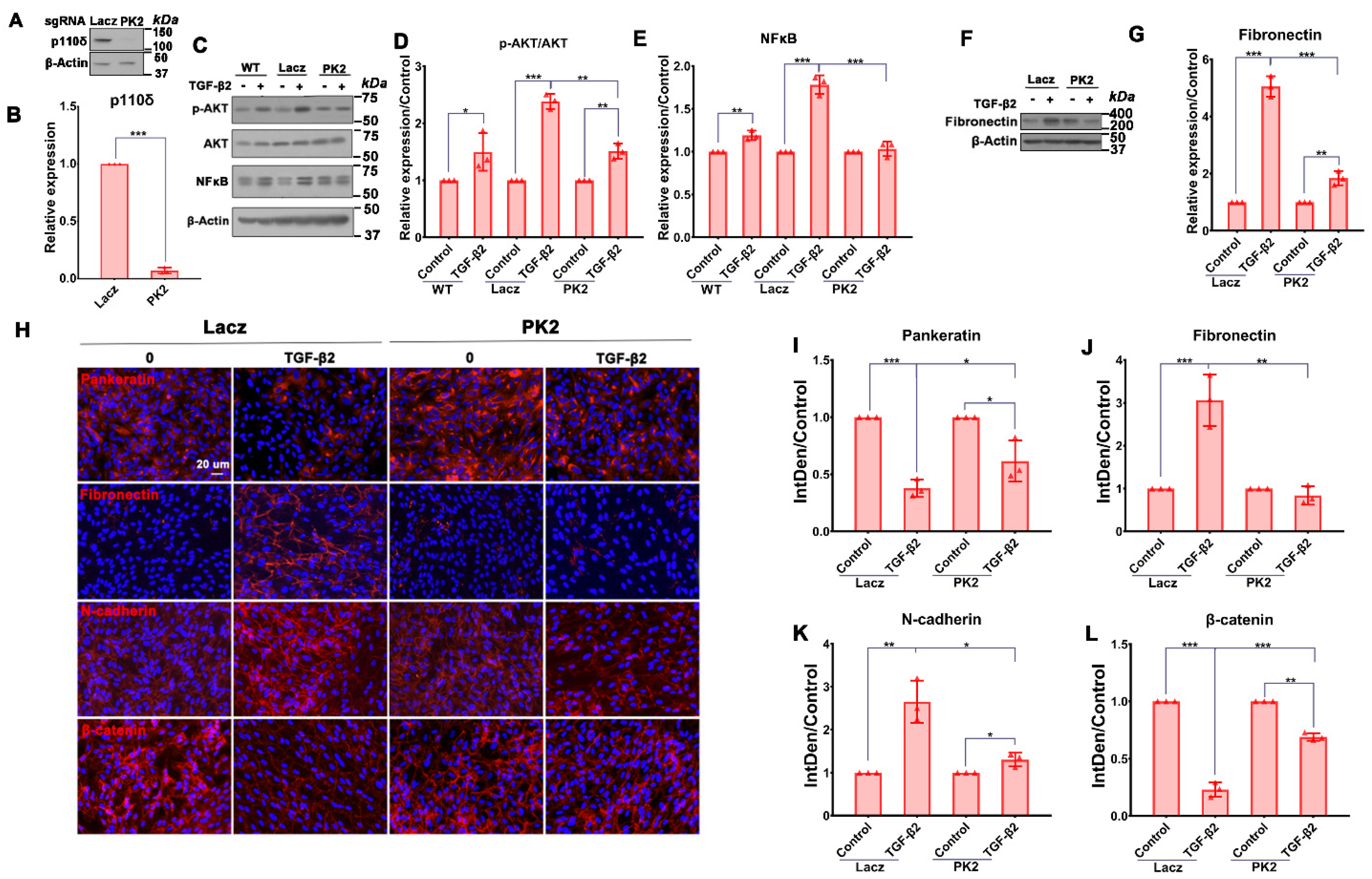
3.3. Depletion of PI3Kδ Blocks TGF-β2-Induced Akt Activation and NFκB/p65 Protein Expression in RPE Cells
3.4. Depletion of PI3Kδ in RPE Cells Prevents TGF-β2-Induced EMT
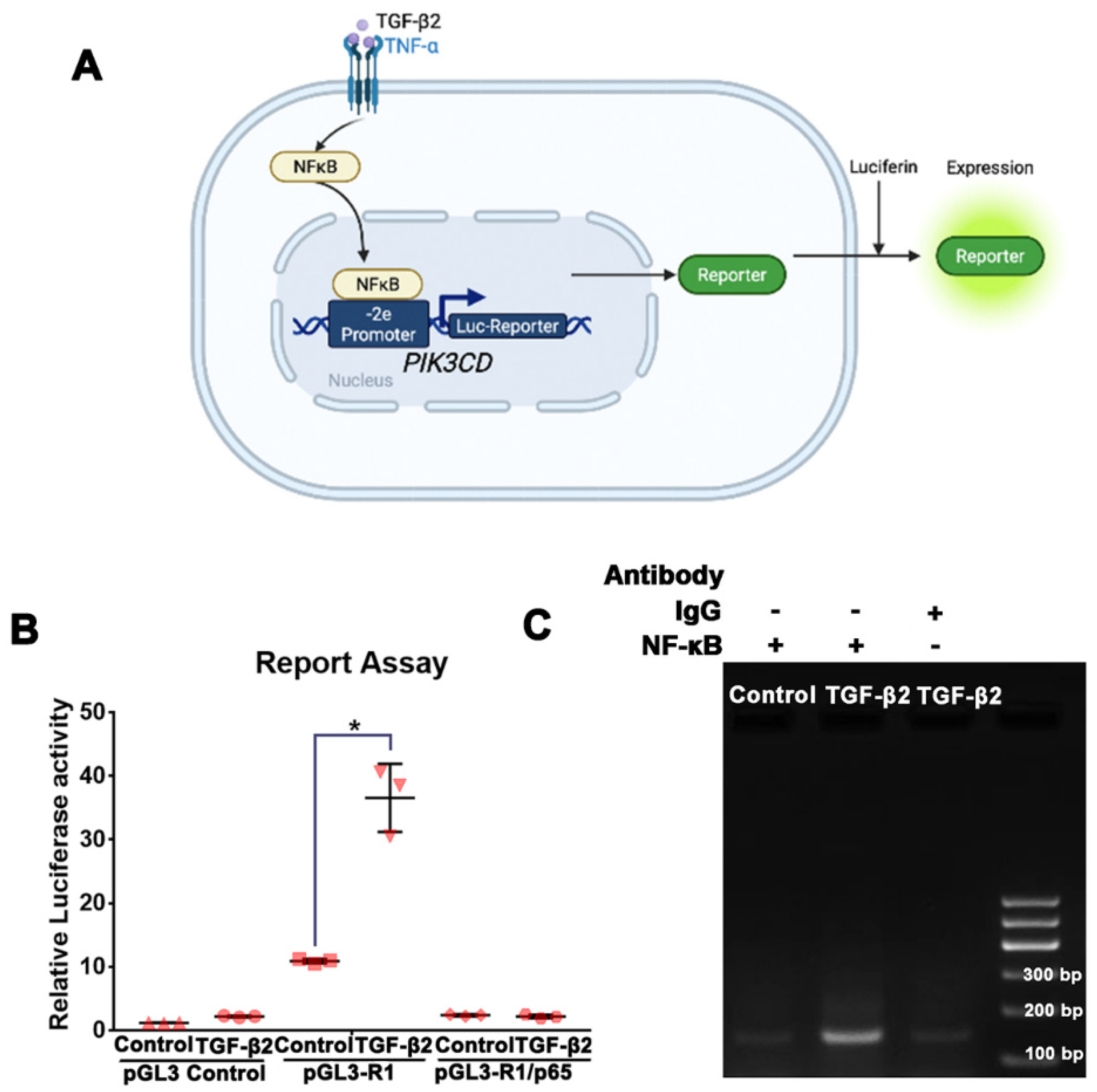
3.5. NFκB/p65 Binding to the PIK3CD-2e Promoter Is Required for the TGF-β2-Induced p110δ Expression in RPE Cells
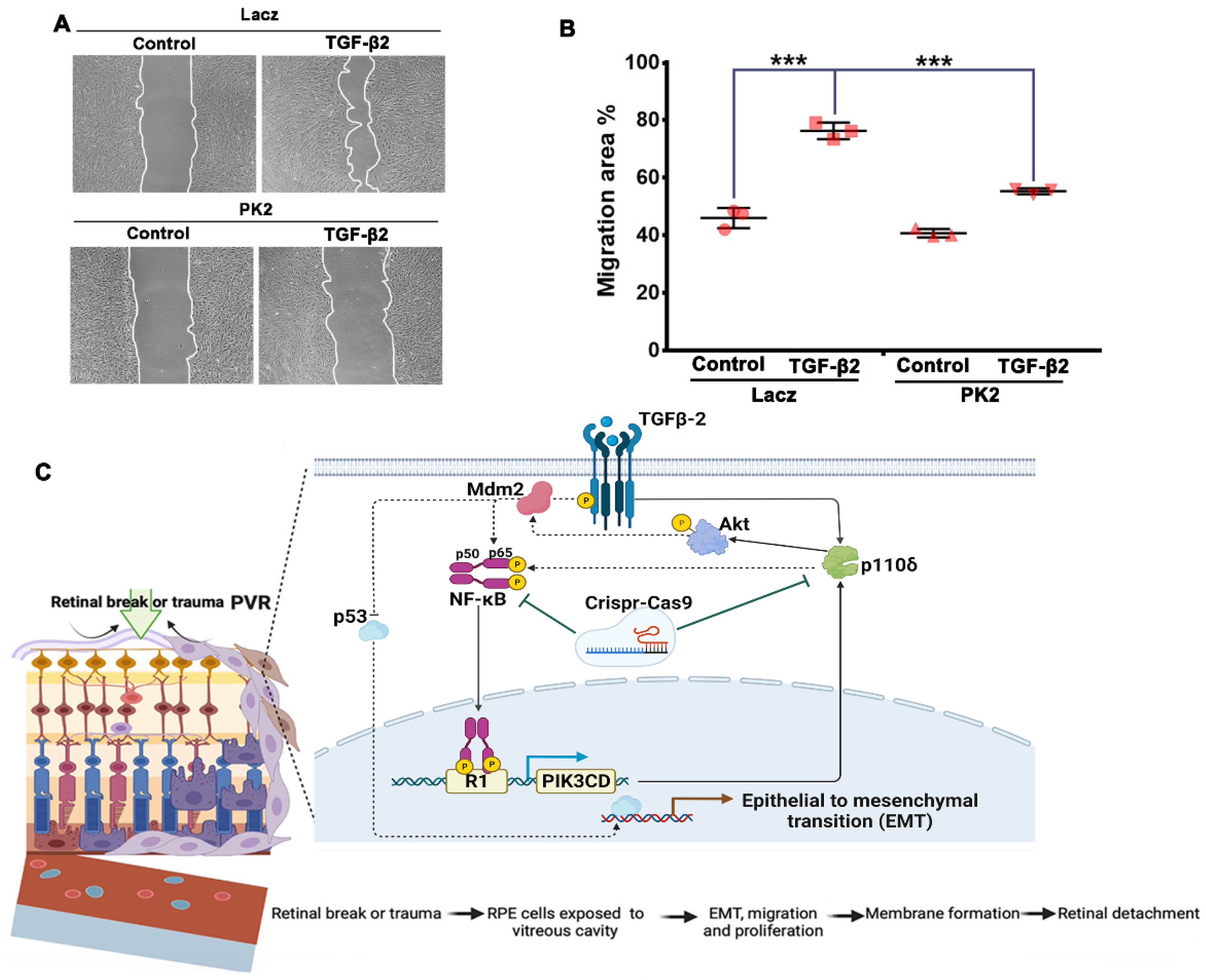
3.6. Depletion of PI3Kδ Prevents TGF-β2-Induced Migration of RPE Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campochiaro, P.A. Pathogenic mechanisms in proliferative vitreoretinopathy. Arch. Ophthalmol. 1997, 115, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.N.; He, S.; Spee, C.; Cui, J.Z.; Ryan, S.J. Hinton DR. In vivo models of proliferative vitreoretinopathy. Nat. Protoc. 2007, 2, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Pastor, J.C.; Rojas, J.; Pastor-Idoate, S.; Di-Lauro, S.; Gonzalez-Buendia, L.; Delgado-Tirado, S. Proliferative vitreoretinopathy: A new concept of disease pathogenesis and practical consequences. Prog. Retin. Eye Res. 2016, 51, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Zandi, S.; Pfister, I.B.; Traine, P.G.; Tappeiner, C.; Despont, A.; Rieben, R.; Skowronska, M.; Garweg, J.G. Biomarkers for PVR in rhegmatogenous retinal detachment. PLoS ONE 2019, 14, e0214674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, L.; Boles, N.C.; Fernandes, M.; Nachmani, B.; Gentile, R.; Blenkinsop, T.A. P38 inhibition reverses TGFbeta1 and TNFalpha-induced contraction in a model of proliferative vitreoretinopathy. Commun. Biol. 2019, 2, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, G.W.; Broman, A.T.; Hindman, H.B.; Grant, M.P. Vision survival after open globe injury predicted by classification and regression tree analysis. Ophthalmology 2018, 115, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Weichel, E.D.; Colyer, M.H.; Ludlow, S.E.; Bower, K.S.; Eiseman, A.S. Combat ocular trauma visual outcomes during operations iraqi and enduring freedom. Ophthalmology 2018, 115, 2235–2245. [Google Scholar] [CrossRef]
- Erdurman, F.C.; Hurmeric, V.; Gokce, G.; Durukan, A.H.; Sobaci, G.; Altinsoy, H.I. Ocular injuries from improvised explosive devices. Eye 2011, 25, 1491–1498. [Google Scholar] [CrossRef]
- Zhou, Q.; Xu, G.; Zhang, X.; Cao, C.; Zhou, Z. Proteomics of post-traumatic proliferative vitreoretinopathy in rabbit retina reveals alterations to a variety of functional proteins. Curr. Eye Res. 2012, 37, 318–326. [Google Scholar] [CrossRef]
- Morescalchi, F.; Duse, S.; Gambicorti, E.; Romano, M.R.; Costagliola, C.; Semeraro, F. Proliferative vitreoretinopathy after eye injuries: An overexpression of growth factors and cytokines leading to a retinal keloid. Mediat. Inflamm. 2013, 2013, 269787. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, H.; Xu, X.; Hu, Y.; Wang, C.; Ma, Z. Traumatic proliferative vitreoretinopathy: Clinical and Histopathological Observations. Retina 2017, 37, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Abdullatif, A.M.; Macky, T.A.; Abdullatif, M.M.; Nassar, K.; Grisanti, S.; Mortada, H.A.; Soliman, M.M. Intravitreal decorin preventing proliferative vitreoretinopathy in perforating injuries: A pilot study. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 2473–2481. [Google Scholar] [CrossRef] [PubMed]
- Justin, G.A.; Baker, K.M.; Brooks, D.I.; Ryan, D.S.; Weichel, E.D.; Colyer, M.H. Intraocular Foreign Body Trauma in Operation Iraqi Freedom and Operation Enduring Freedom: 2001 to 2011. Ophthalmology 2018, 125, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Pennock, S.; Kim, D.; Mukai, S.; Kuhnle, M.; Chun, D.W.; Matsubara, J.; Cui, J.; Ma, P.; Maberley, D.; Samad, A.; et al. Ranibizumab is a potential prophylaxis for proliferative vitreoretinopathy, a nonangiogenic blinding disease. Am. J. Pathol. 2013, 182, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Negrel, A.D.; Thylefors, B. The global impact of eye injuries. Ophthalmic Epidemiol. 1998, 5, 143–169. [Google Scholar] [CrossRef]
- Asaria, R.H.; Charteris, D.G. Proliferative vitreoretinopathy: Developments in pathogenesis and treatment. Compr. Ophthalmol. Update 2006, 7, 179–185. [Google Scholar]
- Di Lauro, S.; Kadhim, M.R.; Charteris, D.G.; Pastor, J.C. Classifications for Proliferative Vitreoretinopathy (PVR): An Analysis of Their Use in Publications over the Last 15 Years. J. Ophthalmol. 2016, 2016, 7807596. [Google Scholar] [CrossRef] [Green Version]
- Assi, A.; Khoueir, Z.; Helou, C.; Fakhoury, H.; Cherfan, G. Intraocular application of Mitomycin C to prevent proliferative vitreoretinopathy in perforating and severe intraocular foreign body injuries. Eye 2019, 33, 1261–1270. [Google Scholar] [CrossRef]
- Wubben, T.J.; Besirli, C.G.; Zacks, D.N. Pharmacotherapies for Retinal Detachment. Ophthalmology 2016, 123, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Sadaka, A.; Giuliari, G.P. Proliferative vitreoretinopathy: Current and emerging treatments. Clin. Ophthalmol. 2012, 6, 1325–1333. [Google Scholar]
- Zhou, G.; Duan, Y.; Ma, G.; Wu, W.; Hu, Z.; Chen, N.; Chee, Y.; Cui, J.; Samad, A.; Matsubara, J.A.; et al. Introduction of the MDM2 T309G Mutation in Primary Human Retinal Epithelial Cells Enhances Experimental Proliferative Vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5361–5367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.H.; Strohbehn, A.; Stryjewski, T.; Brodowska, K.; Flamme-Wiese, M.J.; Mullins, R.F.; Eliott, D. Posteriorly inserted vitreous base: Preoperative Characteristics, Intraoperative Findings, and Outcomes After Vitrectomy. Retina 2020, 40, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Ma, J.X.; Shang, Q.L.; An, J.B.; Chen, H.T.; Wang, C.X. Safety, pharmacokinetics, and prevention effect of intraocular crocetin in proliferative vitreoretinopathy. Biomed. Pharmacother. 2019, 109, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Pennock, S.; Haddock, L.J.; Eliott, D.; Mukai, S.; Kazlauskas, A. Is neutralizing vitreal growth factors a viable strategy to prevent proliferative vitreoretinopathy? Prog. Retin. Eye Res. 2014, 40, 16–34. [Google Scholar] [CrossRef]
- Tamiya, S.; Kaplan, H.J. Role of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Exp. Eye Res. 2016, 142, 26–31. [Google Scholar] [CrossRef]
- He, H.; Kuriyan, A.E.; Su, C.W.; Mahabole, M.; Zhang, Y.; Zhu, Y.T.; Flynn, H.W.; Parel, J.M.; Tseng, S.C. Inhibition of Proliferation and Epithelial Mesenchymal Transition in Retinal Pigment Epithelial Cells by Heavy Chain-Hyaluronan/Pentraxin 3. Sci. Rep. 2017, 7, 43736. [Google Scholar] [CrossRef]
- Ikuno, Y.; Leong, F.L.; Kazlauskas, A. PI3K and PLCgamma play a central role in experimental PVR. Investig. Ophthalmol. Vis. Sci. 2002, 43, 483–489. [Google Scholar]
- Lei, H.; Velez, G.; Kazlauskas, A. Pathological signaling via platelet-derived growth factor receptor {alpha}of involves chronic activation of Akt and suppression of p53. Mol. Cell. Biol. 2001, 31, 1788–1799. [Google Scholar] [CrossRef] [Green Version]
- Moysidis, S.N.; Thanos, A.; Vavvas, D.G. Mechanisms of inflammation in proliferative vitreoretinopathy: From bench to bedside. Mediat. Inflamm. 2012, 2012, 815937. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, M.A.; Bombardieri, M.; Pitzalis, C.; Vanhaesebroeck, B. Isoform-selective induction of human p110delta PI3K expression by TNFalpha: Identification of a new and inducible PIK3CD promoter. Biochem. J. 2012, 443, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Zhou, G.; Han, H.; Huang, X.; Jiang, H.; Mukai, S.; Kazlauskas, A.; Cui, J.; Matsubara, J.A.; Vanhaesebroeck, B.; et al. PI3Kdelta as a Novel Therapeutic Target in Pathological Angiogenesis. Diabetes 2020, 69, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Chen, N.; Huang, X.; Liu, B.; Tian, J.; Lei, H. Phosphoinositide 3-kinase delta inactivation prevents vitreous-induced activation of AKT/MDM2/p53 and migration of retinal pigment epithelial cells. J. Biol. Chem. 2019, 294, 15408–15417. [Google Scholar] [CrossRef]
- Liu, B.; Song, J.; Han, H.; Hu, Z.; Chen, N.; Cui, J.; Matsubara, J.A.; Zhong, J.; Lei, H. Blockade of MDM2 with inactive Cas9 prevents epithelial to mesenchymal transition in retinal pigment epithelial cells. Lab. Investig. 2019, 99, 1874–1886. [Google Scholar] [CrossRef]
- Connor, T.B.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M. Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J. Clin. Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef]
- Schütze, S.; Potthoff, K.; Machleidt, T.; Berkovic, D.; Wiegmann, K.; Krönke, M. TNF activates NF-KB by phosphatidylcholine-specific sphingomyelin breakdown. Cell 1992, 71, 765–776. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Dijke, P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.I.; Heldin, C.H.; Miyazono, K.; et al. TGF-beta receptor-mediated signaling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [Green Version]
- Majzoub, R.N.; Chan, C.L.; Ewert, K.K.; Silva, B.F.; Liang, K.S.; Safinya, C.R. Fluorescence microscopy colocalization of lipid-nucleic acid nanoparticles with wildtype and mutant Rab5-GFP: A platform for investigating early endosomal events. Biochim. Biophys. Acta 2015, 1848, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Li, H.; Li, M.; Wang, F. Mechanisms of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Discov. Med. 2015, 20, 207–217. [Google Scholar] [PubMed]
- Mudhar, H.S. A brief review of the histopathology of proliferative vitreoretinopathy (PVR). Eye 2020, 34, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Rhéaume, M.A.; Velez, G.; Mukai, S.; Kazlauskas, A. Expression of PDGFR {alpha} Is a Determinant of the PVR Potential of ARPE19 Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5016–5021. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Rheaume, M.A.; Cui, J.; Mukai, S.; Maberley, D.; Samad, A.; Matsubara, J.; Kazlauskas, A. A novel function of p53: A gatekeeper of retinal detachment. Am. J. Pathol. 2012, 181, 866–874. [Google Scholar] [CrossRef] [Green Version]
- Lei, H.; Kazlauskas, A. Growth factors outside of the PDGF family employ ROS/SFKs to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J. Biol. Chem. 2009, 284, 6329–6336. [Google Scholar] [CrossRef] [Green Version]
- Frick, C.L.; Yarka, C.; Nunns, H.; Goentoro, L. Sensing relative signal in the Tgf-beta/Smad pathway. Proc. Natl. Acad. Sci. USA 2017, 114, E2975–E2982. [Google Scholar] [CrossRef] [Green Version]
- Araki, S.; Eitel, J.A.; Batuello, C.N.; Bijangi-Vishehsaraei, K.; Xie, X.J.; Danielpour, D.; Pollok, K.E.; Boothman, D.A.; Mayo, L.D. TGF-beta1-induced expression of human Mdm2 correlates with late-stage metastatic breast cancer. J. Clin. Investig. 2010, 120, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, T.; Otsuka, Y.; Sabe, H. p53-Dependent and -Independent Epithelial Integrity: Beyond miRNAs and Metabolic Fluctuations. Cancers 2018, 10, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Widen, S.G.; Yang, J.; Wood, T.G.; Kudlicki, A.; Zhao, Y.; Brasier, A.R. The NFkappaB subunit RELA is a master transcriptional regulator of the committed epithelial-mesenchymal transition in airway epithelial cells. J. Biol. Chem. 2018, 293, 16528–16545. [Google Scholar] [CrossRef]
- Lei, H.; Kazlauskas, A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor alpha. Mol. Cell. Biol. 2014, 34, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Whitehead, M.A.; Pineiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. 2016, 94, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Idoate, S.; Rodriguez-Hernandez, I.; Rojas, J.; Fernandez, I.; Garcia-Gutierrez, M.T.; Ruiz-Moreno, J.M.; Rocha-Sousa, A.; Ramkissoon, Y.; Harsum, S.; MacLaren, R.E.; et al. The T309G MDM2 gene polymorphism is a novel risk factor for proliferative vitreoretinopathy. PLoS ONE 2013, 8, e82283. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Ma, G.; Huang, X.; D’Amore, P.A.; Zhang, F.; Lei, H. The Clustered, Regularly Interspaced, Short Palindromic Repeats-associated Endonuclease 9 (CRISPR/Cas9)-created MDM2 T309G Mutation Enhances Vitreous-induced Expression of MDM2 and Proliferation and Survival of Cells. J. Biol. Chem. 2016, 291, 16339–16347. [Google Scholar] [CrossRef] [Green Version]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Post, S.M.; Quintás-Cardama, A.; Pant, V.; Iwakuma, T.; Hamir, A.; Jackson, J.G.; Maccio, D.R.; Bond, G.L.; Johnson, D.G.; Levine, A.J.; et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell 2010, 18, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Findley, H.W.; Zhou, M. MDM2 induces NF-kappaB/p65 expression transcriptionally through Sp1-binding sites: A novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood 2002, 99, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.; Nudleman, E.; Weinreb, R.N. Animal Models of Proliferative Vitreoretinopathy and Their Use in Pharmaceutical Investigations. Ophthalmic Res. 2018, 60, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. A Development of a gene-editing. approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Xin, T.; Han, H.; Wu, W.; Huang, X.; Cui, J.; Matsubara, J.A.; Song, J.; Wang, F.; Colyer, M.; Lei, H. Idelalisib inhibits vitreous-induced Akt activation and proliferation of retinal pigment epithelial cells from epiretinal membranes. Exp. Eye Res. 2020, 190, 107884. [Google Scholar] [CrossRef] [PubMed]
- Idrees, S.; Sridhar, J.; Kuriyan, A.E. Proliferative Vitreoretinopathy: A Review. Int. Ophthalmol. Clin. 2019, 59, 221–240. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.; Yang, Y.; Han, Z.; Wang, L.; Dong, L.; Qi, H.; Liu, B.; Tian, J.; Vanhaesebroeck, B.; Kazlauskas, A.; et al. NFκB-Mediated Expression of Phosphoinositide 3-Kinase δ Is Critical for Mesenchymal Transition in Retinal Pigment Epithelial Cells. Cells 2023, 12, 207. https://doi.org/10.3390/cells12020207
Han H, Yang Y, Han Z, Wang L, Dong L, Qi H, Liu B, Tian J, Vanhaesebroeck B, Kazlauskas A, et al. NFκB-Mediated Expression of Phosphoinositide 3-Kinase δ Is Critical for Mesenchymal Transition in Retinal Pigment Epithelial Cells. Cells. 2023; 12(2):207. https://doi.org/10.3390/cells12020207
Chicago/Turabian StyleHan, Haote, Yanhui Yang, Zhuo Han, Luping Wang, Lijun Dong, Hui Qi, Bing Liu, Jingkui Tian, Bart Vanhaesebroeck, Andrius Kazlauskas, and et al. 2023. "NFκB-Mediated Expression of Phosphoinositide 3-Kinase δ Is Critical for Mesenchymal Transition in Retinal Pigment Epithelial Cells" Cells 12, no. 2: 207. https://doi.org/10.3390/cells12020207