
Linking Mechanisms of Vitamin D Signaling with Multiple Sclerosis


Abstract
:1. Introduction
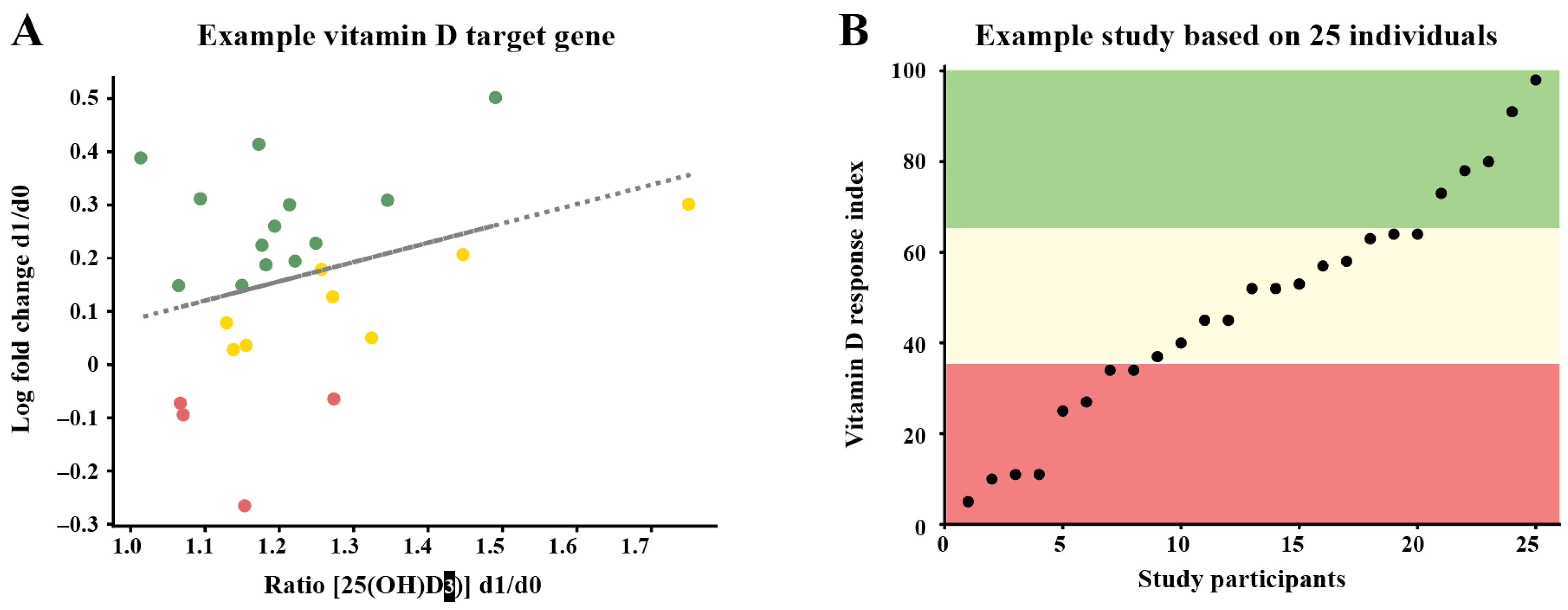
2. Vitamin D Status versus Responsiveness
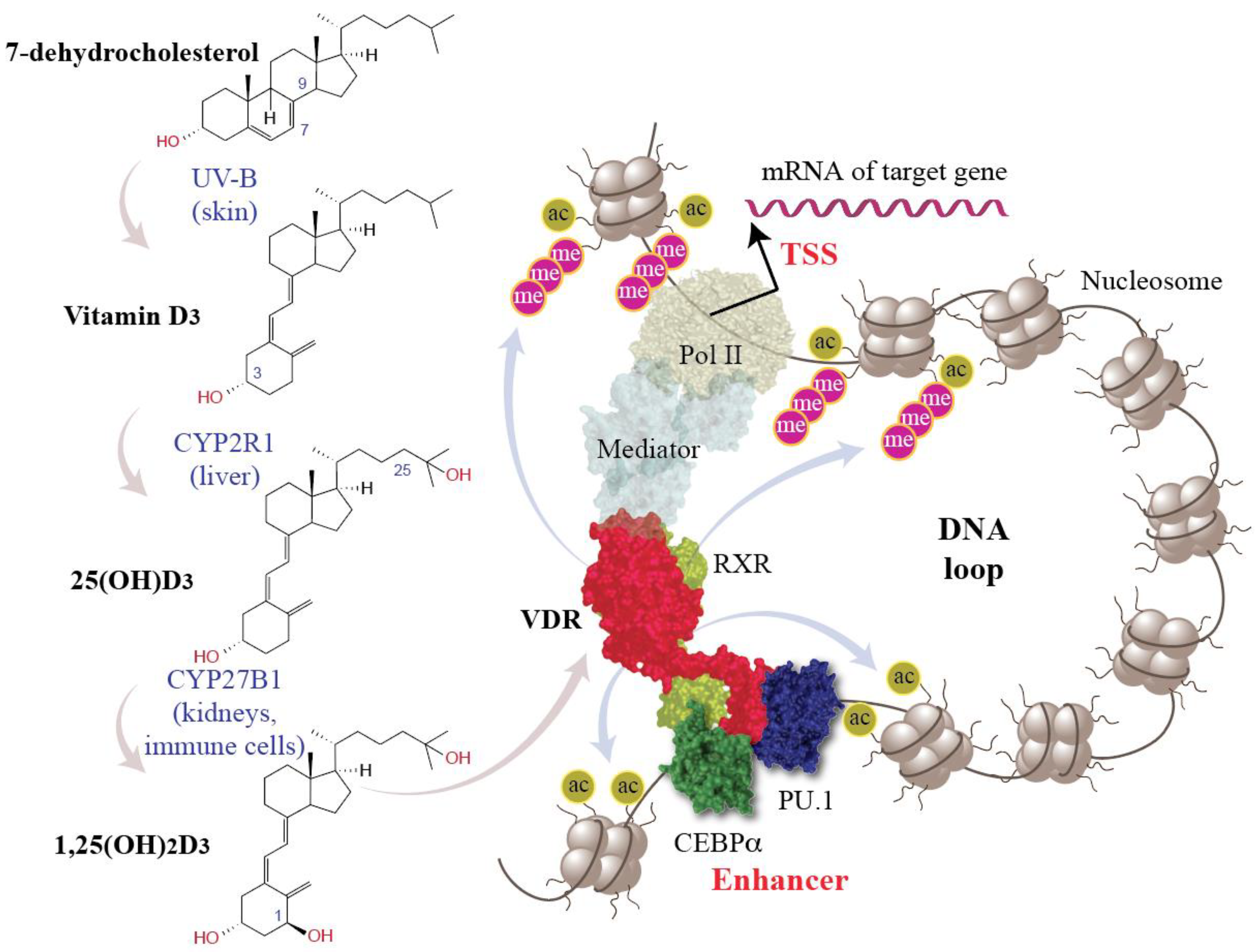
3. Vitamin D Regulates the Epigenome and Transcriptome of Immune Cells
4. Vitamin D Target Genes with Impact for MS
5. Relating MS Pathogenesis to Vitamin D
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Charabati, M.; Wheeler, M.A.; Weiner, H.L.; Quintana, F.J. Multiple sclerosis: Neuroimmune crosstalk and therapeutic targeting. Cell 2023, 186, 1309–1327. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- O’Quinn, D.B.; Palmer, M.T.; Lee, Y.K.; Weaver, C.T. Emergence of the Th17 pathway and its role in host defense. Adv. Immunol. 2008, 99, 115–163. [Google Scholar] [CrossRef]
- Bierhansl, L.; Hartung, H.P.; Aktas, O.; Ruck, T.; Roden, M.; Meuth, S.G. Thinking outside the box: Non-canonical targets in multiple sclerosis. Nat. Rev. Drug Discov. 2022, 21, 578–600. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Velleuer, E.; Molnar, F. Molecular Medicine: How Science Works; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Ordovas, J.M.; Ferguson, L.R.; Tai, E.S.; Mathers, J.C. Personalised nutrition and health. BMJ 2018, 361, bmj.k2173. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics, C. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Maugeri, N.J.; Handunnetthi, L.; Lincoln, M.R.; Orton, S.M.; Dyment, D.A.; Deluca, G.C.; Herrera, B.M.; Chao, M.J.; Sadovnick, A.D.; et al. Expression of the multiple sclerosis-associated MHC class II allele HLA-DRB1*1501 is regulated by vitamin D. PLoS Genet. 2009, 5, e1000369. [Google Scholar] [CrossRef]
- Wang, J.; Jelcic, I.; Muhlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20. [Google Scholar] [CrossRef]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef]
- Hedstrom, A.K.; Olsson, T.; Kockum, I.; Hillert, J.; Alfredsson, L. Low sun exposure increases multiple sclerosis risk both directly and indirectly. J. Neurol. 2020, 267, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Ostkamp, P.; Salmen, A.; Pignolet, B.; Gorlich, D.; Andlauer, T.F.M.; Schulte-Mecklenbeck, A.; Gonzalez-Escamilla, G.; Bucciarelli, F.; Gennero, I.; Breuer, J.; et al. Sunlight exposure exerts immunomodulatory effects to reduce multiple sclerosis severity. Proc. Natl. Acad. Sci. USA 2021, 118, e2018457118. [Google Scholar] [CrossRef] [PubMed]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and multiple sclerosis: A comprehensive review. Neurol. Ther. 2018, 7, 59–85. [Google Scholar] [CrossRef]
- Pierrot-Deseilligny, C.; Souberbielle, J.C. Vitamin D and multiple sclerosis: An update. Mult. Scler. Relat. Disord. 2017, 14, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bendik, I.; Friedel, A.; Roos, F.F.; Weber, P.; Eggersdorfer, M. Vitamin D: A critical and essential micronutrient for human health. Front. Physiol. 2014, 5, 248. [Google Scholar] [CrossRef]
- Holick, M.F. Photobiology of vitamin D. In Vitamin D, 3rd ed.; Academic Press: Cambridge, MA, USA, 2011; pp. 13–22. [Google Scholar] [CrossRef]
- Pierrot-Deseilligny, C.; Souberbielle, J.C. Is hypovitaminosis D one of the environmental risk factors for multiple sclerosis? Brain A J. Neurol. 2010, 133, 1869–1888. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S., Jr.; Blizzard, L.; Otahal, P.; Van der Mei, I.; Taylor, B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1132–1141. [Google Scholar] [CrossRef]
- van de Peppel, J.; van Leeuwen, J.P. Vitamin D and gene networks in human osteoblasts. Front. Physiol. 2014, 5, 137. [Google Scholar] [CrossRef]
- Carmeliet, G.; Dermauw, V.; Bouillon, R. Vitamin D signaling in calcium and bone homeostasis: A delicate balance. Best Pract. Research. Clin. Endocrinol. Metab. 2015, 29, 621–631. [Google Scholar] [CrossRef]
- Chun, R.F.; Liu, P.T.; Modlin, R.L.; Adams, J.S.; Hewison, M. Impact of vitamin D on immune function: Lessons learned from genome-wide analysis. Front. Physiol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Hanel, A.; Carlberg, C. Time-resolved gene expression analysis monitors the regulation of inflammatory mediators and attenuation of adaptive immune response by vitamin D. Int. J. Mol. Sci. 2022, 23, 911. [Google Scholar] [CrossRef]
- Malmberg, H.R.; Hanel, A.; Taipale, M.; Heikkinen, S.; Carlberg, C. Vitamin D treatment sequence is critical for transcriptome modulation of immune challenged primary human cells. Front. Immunol. 2021, 12, 754056. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; McComish, B.J.; Burdon, K.P.; Taylor, B.V.; Körner, H. The association between vitamin D and multiple sclerosis risk: 1,25(OH)2D3 induces super-enhancers bound by VDR. Front. Immunol. 2019, 10, 488. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, L.E.; Raza, K.; Hewison, M. Vitamin D in rheumatoid arthritis-towards clinical application. Nat. Rev. Rheumatol. 2016, 12, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.; Cooper, S.C.; Ghosh, S.; Hewison, M. The role of vitamin D in Inflammatory bowel disease: Mechanism to management. Nutrients 2019, 11, 1019. [Google Scholar] [CrossRef] [PubMed]
- Infante, M.; Ricordi, C.; Sanchez, J.; Clare-Salzler, M.J.; Padilla, N.; Fuenmayor, V.; Chavez, C.; Alvarez, A.; Baidal, D.; Alejandro, R.; et al. Influence of vitamin D on islet autoimmunity and beta-cell function in type 1 diabetes. Nutrients 2019, 11, 2185. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Cook, N.R.; Alexander, E.K.; Friedman, S.; Walter, J.; Bubes, V.; Kotler, G.; Lee, I.M.; Manson, J.E.; Costenbader, K.H. Vitamin D and marine omega 3 fatty acid supplementation and incident autoimmune disease: VITAL randomized controlled trial. BMJ 2022, 376, e066452. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D supplements and prevention of cancer and cardiovascular disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef]
- Bouillon, R.; Manousaki, D.; Rosen, C.; Trajanoska, K.; Rivadeneira, F.; Richards, J.B. The health effects of vitamin D supplementation: Evidence from human studies. Nat. Rev. Endocrinol. 2022, 18, 96–110. [Google Scholar] [CrossRef]
- Hollis, B.W. Circulating 25-hydroxyvitamin D levels indicative of vitamin D sufficiency: Implications for establishing a new effective dietary intake recommendation for vitamin D. J. Nutr. 2005, 135, 317–322. [Google Scholar] [CrossRef]
- Del Valle, H.B.; Yaktine, A.L.; Taylor, C.L.; Ross, A.C. (Eds.) Dietary Reference Intakes for Calcium and Vitamin D; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from Vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [PubMed]
- Pludowski, P.; Holick, M.F.; Pilz, S.; Wagner, C.L.; Hollis, B.W.; Grant, W.B.; Shoenfeld, Y.; Lerchbaum, E.; Llewellyn, D.J.; Kienreich, K.; et al. Vitamin D effects on musculoskeletal health, immunity, autoimmunity, cardiovascular disease, cancer, fertility, pregnancy, dementia and mortality-a review of recent evidence. Autoimmun. Rev. 2013, 12, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M.; Endocrine, S. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef]
- Genomes Project, C.; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Carlberg, C.; Seuter, S.; de Mello, V.D.; Schwab, U.; Voutilainen, S.; Pulkki, K.; Nurmi, T.; Virtanen, J.; Tuomainen, T.P.; Uusitupa, M. Primary Vitamin D target genes allow a categorization of possible benefits of vitamin D3 supplementation. PLoS ONE 2013, 8, e71042. [Google Scholar] [CrossRef] [PubMed]
- Wilfinger, J.; Seuter, S.; Tuomainen, T.-P.; Virtanen, J.K.; Voutilainen, S.; Nurmi, T.; de Mello, V.D.F.; Uusitupa, M.; Carlberg, C. Primary vitamin D receptor target genes as biomarkers for the vitamin D3 status in the hematopoietic system. J. Nutr. Biochem. 2014, 25, 875–884. [Google Scholar] [CrossRef]
- Ryynänen, J.; Neme, A.; Tuomainen, T.P.; Virtanen, J.K.; Voutilainen, S.; Nurmi, T.; de Mello, V.D.; Uusitupa, M.; Carlberg, C. Changes in vitamin D target gene expression in adipose tissue monitor the vitamin D response of human individuals. Mol. Nutr. Food Res. 2014, 58, 2036–2045. [Google Scholar] [CrossRef]
- Saksa, N.; Neme, A.; Ryynänen, J.; Uusitupa, M.; de Mello, V.D.; Voutilainen, S.; Nurmi, T.; Virtanen, J.K.; Tuomainen, T.P.; Carlberg, C. Dissecting high from low responders in a vitamin D3 intervention study. J. Steroid Biochem. Mol. Biol. 2015, 148, 275–282. [Google Scholar] [CrossRef]
- Vukic, M.; Neme, A.; Seuter, S.; Saksa, N.; de Mello, V.D.; Nurmi, T.; Uusitupa, M.; Tuomainen, T.P.; Virtanen, J.K.; Carlberg, C. Relevance of vitamin D receptor target genes for monitoring the Vitamin D responsiveness of primary human cells. PLoS ONE 2015, 10, e0124339. [Google Scholar] [CrossRef]
- Seuter, S.; Virtanen, J.K.; Nurmi, T.; Pihlajamäki, J.; Mursu, J.; Voutilainen, S.; Tuomainen, T.P.; Neme, A.; Carlberg, C. Molecular evaluation of vitamin D responsiveness of healthy young adults. J. Steroid Biochem. Mol. Biol. 2017, 174, 314–321. [Google Scholar] [CrossRef]
- Carlberg, C. Molecular approaches for optimizing Vitamin D supplementation. Vitam. Horm. 2016, 100, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Haq, A. The concept of the personal vitamin D response index. J. Steroid Biochem. Mol. Biol. 2018, 175, 12–17. [Google Scholar] [CrossRef]
- Salzer, J.; Hallmans, G.; Nystrom, M.; Stenlund, H.; Wadell, G.; Sundstrom, P. Vitamin D as a protective factor in multiple sclerosis. Neurology 2012, 79, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Mangin, M.; Sinha, R.; Fincher, K. Inflammation and vitamin D: The infection connection. Inflamm. Res. 2014, 63, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; O’Reilly, P.F.; Aschard, H.; Hsu, Y.H.; Richards, J.B.; Dupuis, J.; Ingelsson, E.; Karasik, D.; Pilz, S.; Berry, D.; et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Trabzuni, D.; Forabosco, P.; Smith, C.; Walker, R.; Dillman, A.; Sveinbjornsdottir, S.; North American Brain Expression Consortium; UK Brain Expression Consortium; Hardy, J.; et al. Genetic evidence for a pathogenic role for the vitamin D3 metabolizing enzyme CYP24A1 in multiple sclerosis. Mult. Scler. Relat. Disord. 2014, 3, 211–219. [Google Scholar] [CrossRef]
- Manousaki, D.; Dudding, T.; Haworth, S.; Hsu, Y.H.; Liu, C.T.; Medina-Gomez, C.; Voortman, T.; van der Velde, N.; Melhus, H.; Robinson-Cohen, C.; et al. Low-frequency synonymous coding variation in CYP2R1 has large efects on vitamin D levels and risk of multiple sclerosis. Am. J. Hum. Genet. 2017, 101, 227–238. [Google Scholar] [CrossRef]
- Alloza, I.; Otaegui, D.; de Lapuente, A.L.; Antiguedad, A.; Varade, J.; Nunez, C.; Arroyo, R.; Urcelay, E.; Fernandez, O.; Leyva, L.; et al. ANKRD55 and DHCR7 are novel multiple sclerosis risk loci. Genes Immun. 2012, 13, 253–257. [Google Scholar] [CrossRef]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef]
- Hanel, A.; Neme, A.; Malinen, M.; Hamalainen, E.; Malmberg, H.R.; Etheve, S.; Tuomainen, T.P.; Virtanen, J.K.; Bendik, I.; Carlberg, C. Common and personal target genes of the micronutrient vitamin D in primary immune cells from human peripheral blood. Sci. Rep. 2020, 10, 21051. [Google Scholar] [CrossRef] [PubMed]
- Hanel, A.; Veldhuizen, C.; Carlberg, C. Gene-regulatory potential of 25-hydroxyvitamin D3 and D2. Front. Nutr. 2022, 9, 910601. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Rando, O.J.; Chang, H.Y. Genome-wide views of chromatin structure. Annu. Rev. Biochem. 2009, 78, 245–271. [Google Scholar] [CrossRef]
- Carlberg, C. Molecular endocrinology of Vitamin D on the epigenome level. Mol. Cell. Endocrinol. 2017, 453, 14–21. [Google Scholar] [CrossRef]
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167. [Google Scholar] [CrossRef]
- Tuoresmäki, P.; Väisänen, S.; Neme, A.; Heikkinen, S.; Carlberg, C. Patterns of genome-wide VDR locations. PLoS ONE 2014, 9, e96105. [Google Scholar] [CrossRef]
- Neme, A.; Seuter, S.; Carlberg, C. Selective regulation of biological processes by vitamin D based on the spatio-temporal cistrome of its receptor. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2017, 1860, 952–961. [Google Scholar] [CrossRef]
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011, 144, 296–309. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Habibi, E.; Wang, S.-Y.; Arts, R.J.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan reverses the epigenetic state of LPS-Induced immunological tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Zhang, S.M.; O’Reilly, E.; Hernan, M.A.; Olek, M.J.; Willett, W.C.; Ascherio, A. Vitamin D intake and incidence of multiple sclerosis. Neurology 2004, 62, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Riise, T.; Bjornevik, K.; Holmoy, T.; Kampman, M.T.; Magalhaes, S.; Pugliatti, M.; Wolfson, C.; Myhr, K.M. Timing of use of cod liver oil, a vitamin D source, and multiple sclerosis risk: The EnvIMS study. Mult. Scler. 2015, 21, 1856–1864. [Google Scholar] [CrossRef]
- Escriva, H.; Bertrand, S.; Laudet, V. The evolution of the nuclear receptor superfamily. Essays Biochem. 2004, 40, 11–26. [Google Scholar] [CrossRef]
- Krasowski, M.D.; Ni, A.; Hagey, L.R.; Ekins, S. Evolution of promiscuous nuclear hormone receptors: LXR, FXR, VDR, PXR, and CAR. Mol. Cell. Endocrinol. 2011, 334, 39–48. [Google Scholar] [CrossRef]
- Heikkinen, S.; Väisänen, S.; Pehkonen, P.; Seuter, S.; Benes, V.; Carlberg, C. Nuclear hormone 1α,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic Acids Res. 2011, 39, 9181–9193. [Google Scholar] [CrossRef]
- Vanherwegen, A.S.; Eelen, G.; Ferreira, G.B.; Ghesquiere, B.; Cook, D.P.; Nikolic, T.; Roep, B.; Carmeliet, P.; Telang, S.; Mathieu, C.; et al. Vitamin D controls the capacity of human dendritic cells to induce functional regulatory T cells by regulation of glucose metabolism. J. Steroid Biochem. Mol. Biol. 2019, 187, 134–145. [Google Scholar] [CrossRef]
- Vanherwegen, A.S.; Gysemans, C.; Mathieu, C. Vitamin D endocrinology on the cross-road between immunity and metabolism. Mol. Cell. Endocrinol. 2017, 453, 52–67. [Google Scholar] [CrossRef]
- Verstuyf, A.; Carmeliet, G.; Bouillon, R.; Mathieu, C. Vitamin D: A pleiotropic hormone. Kidney Int. 2010, 78, 140–145. [Google Scholar] [CrossRef]
- Carlberg, C. The physiology of vitamin D-far more than calcium and bone. Front. Physiol. 2014, 5, 335. [Google Scholar] [CrossRef] [PubMed]
- Barragan, M.; Good, M.; Kolls, J.K. Regulation of dendritic cell function by vitamin D. Nutrients 2015, 7, 8127–8151. [Google Scholar] [CrossRef] [PubMed]
- Dankers, W.; Colin, E.M.; van Hamburg, J.P.; Lubberts, E. Vitamin D in autoimmunity: Molecular mechanisms and therapeutic potential. Front. Immunol. 2016, 7, 697. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. In Seminars in immunology; Academic Press: Cambridge, MA, USA, 2013; Volume 25, pp. 305–312. [Google Scholar] [CrossRef]
- Palmer, M.T.; Lee, Y.K.; Maynard, C.L.; Oliver, J.R.; Bikle, D.D.; Jetten, A.M.; Weaver, C.T. Lineage-specific effects of 1,25-dihydroxyvitamin D3 on the development of effector CD4 T cells. J. Biol. Chem. 2011, 286, 997–1004. [Google Scholar] [CrossRef]
- Jeffery, L.E.; Burke, F.; Mura, M.; Zheng, Y.; Qureshi, O.S.; Hewison, M.; Walker, L.S.; Lammas, D.A.; Raza, K.; Sansom, D.M. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J. Immunol. 2009, 183, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. Jama 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Limketkai, B.N.; Mullin, G.E.; Limsui, D.; Parian, A.M. Role of vitamin D in inflammatory bowel disease. Nutr. Clin. Pract. 2017, 32, 337–345. [Google Scholar] [CrossRef]
- Zeitelhofer, M.; Adzemovic, M.Z.; Gomez-Cabrero, D.; Bergman, P.; Hochmeister, S.; N’Diaye, M.; Paulson, A.; Ruhrmann, S.; Almgren, M.; Tegner, J.N.; et al. Functional genomics analysis of vitamin D effects on CD4+ T cells in vivo in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2017, 114, E1678–E1687. [Google Scholar] [CrossRef]
- Carlberg, C.; Seuter, S.; Nurmi, T.; Tuomainen, T.P.; Virtanen, J.K.; Neme, A. In vivo response of the human epigenome to Vitamin D: A proof-of-principle study. J. Steroid Biochem. Mol. Biol. 2018, 180, 142–148. [Google Scholar] [CrossRef]
- Neme, A.; Seuter, S.; Malinen, M.; Nurmi, T.; Tuomainen, T.P.; Virtanen, J.K.; Carlberg, C. In vivo transcriptome changes of human white blood cells in response to vitamin D. J. Steroid Biochem. Mol. Biol. 2019, 188, 71–76. [Google Scholar] [CrossRef]
- Cortes, M.; Chen, M.J.; Stachura, D.L.; Liu, S.Y.; Kwan, W.; Wright, F.; Vo, L.T.; Theodore, L.N.; Esain, V.; Frost, I.M.; et al. Developmental vitamin D availability impacts hematopoietic stem cell production. Cell Rep. 2016, 17, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, O.; Hanel, A.; Carlberg, C. Key Vitamin D target genes with functions in the immune system. Nutrients 2020, 12, 1140. [Google Scholar] [CrossRef] [PubMed]
- Hanel, A.; Malmberg, H.R.; Carlberg, C. Genome-wide effects of chromatin on Vitamin D signaling. J. Mol. Endocrinol. 2020, 64, R45–R56. [Google Scholar] [CrossRef]
- da Costa, D.S.; Hygino, J.; Ferreira, T.B.; Kasahara, T.M.; Barros, P.O.; Monteiro, C.; Oliveira, A.; Tavares, F.; Vasconcelos, C.C.; Alvarenga, R.; et al. Vitamin D modulates different IL-17-secreting T cell subsets in multiple sclerosis patients. J. Neuroimmunol. 2016, 299, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Muris, A.H.; Smolders, J.; Rolf, L.; Thewissen, M.; Hupperts, R.; Damoiseaux, J.; SOLARIUM Study Group. Immune regulatory effects of high dose vitamin D3 supplementation in a randomized controlled trial in relapsing remitting multiple sclerosis patients receiving IFNbeta; the SOLARIUM study. J. Neuroimmunol. 2016, 300, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Langlois, J.; Denimal, D. Clinical and imaging outcomes after vitamin D supplementation in patients with Multiple Sclerosis: A systematic review. Nutrients 2023, 15, 1945. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vitamin D Response Index | Daily Vitamin D3 Supplementation | Immune System Balance Restored | Clinical Effect |
|---|---|---|---|
| High | Standard (1000 IU) | + | Present |
| Mid | Standard (1000 IU) | +/− | Partial |
| Mid | Mid (2000 IU) | + | Present |
| Low | Standard (1000 IU) | − | Absent |
| Low | High (4000 IU) | + | Present |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlberg, C.; Mycko, M.P. Linking Mechanisms of Vitamin D Signaling with Multiple Sclerosis. Cells 2023, 12, 2391. https://doi.org/10.3390/cells12192391
Carlberg C, Mycko MP. Linking Mechanisms of Vitamin D Signaling with Multiple Sclerosis. Cells. 2023; 12(19):2391. https://doi.org/10.3390/cells12192391
Chicago/Turabian StyleCarlberg, Carsten, and Marcin P. Mycko. 2023. "Linking Mechanisms of Vitamin D Signaling with Multiple Sclerosis" Cells 12, no. 19: 2391. https://doi.org/10.3390/cells12192391