
Knock Out of CGN and CGNL1 in MDCK Cells Affects Claudin-2 but Has a Minor Impact on Tight Junction Barrier Function



Abstract
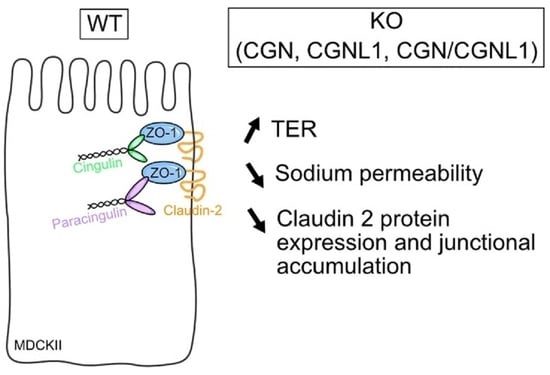
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Measurement of Transepithelial Resistance (TER) and Paracellular Flux Assay
2.3. Dilution Potential of NaCl
2.4. RNASeq Analysis
2.5. Immunoblot Analysis
2.6. Plasmids
2.7. Immunofluoresence Anaylsis
2.8. Quantification and Statistical Analysis
3. Results
3.1. MDCK Cells KO for Either CGN, CGNL1 or Both Show Slightly Increased Transepithelial Resistance but No Change in Permeability to Dextran
3.2. KO of Either CGN or CGNL1 or Both Results in Decreased Sodium Permeability of MDCK Monolayers
3.3. MDCK Cells KO for Either CGN or CGNL1 or Both Show Decreased Claudin-2 Protein Expression
3.4. MDCK Cells KO for CGN, CGNL1, or Both Show Decreased Claudin-2 Accumulation at Junctions

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Rouaud, F.; Sluysmans, S.; Flinois, A.; Shah, J.; Vasileva, E.; Citi, S. Scaffolding proteins of vertebrate apical junctions: Structure, functions and biophysics. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183399. [Google Scholar] [CrossRef]
- Otani, T.; Furuse, M. Tight Junction Structure and Function Revisited. Trends Cell Biol. 2020, 30, 815–817. [Google Scholar] [CrossRef]
- Citi, S.; Guerrera, D.; Spadaro, D.; Shah, J. Epithelial junctions and Rho family GTPases: The zonular signalosome. Small GTPases 2014, 5, e973760. [Google Scholar] [CrossRef] [Green Version]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight junction pore and leak pathways: A dynamic duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [Green Version]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 2006, 68, 403–429. [Google Scholar] [CrossRef]
- Rosenthal, R.; Gunzel, D.; Krug, S.M.; Schulzke, J.D.; Fromm, M.; Yu, A.S. Claudin-2-mediated cation and water transport share a common pore. Acta. Physiol. 2017, 219, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Cordenonsi, M.; D’Atri, F.; Hammar, E.; Parry, D.A.D.; Kendrick-Jones, J.; Shore, D.; Citi, S. Cingulin Contains Globular and Coiled-coil Domains and Interacts with ZO-1, ZO-2, ZO-3, and Myosin. J. Cell Biol. 1999, 147, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, F.; Nadalutti, F.; Citi, S. Evidence for a functional interaction between cingulin and ZO-1 in cultured cells. J. Biol. Chem. 2002, 277, 27757–27764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnishi, H.; Nakahara, T.; Furuse, K.; Sasaki, H.; Tsukita, S.; Furuse, M. JACOP, a novel plaque protein localizing at the apical junctional complex with sequence similarity to cingulin. J. Biol. Chem. 2004, 279, 46014–46022. [Google Scholar] [CrossRef] [Green Version]
- Umeda, K.; Ikenouchi, J.; Katahira-Tayama, S.; Furuse, K.; Sasaki, H.; Nakayama, M.; Matsui, T.; Tsukita, S.; Furuse, M.; Tsukita, S. ZO-1 and ZO-2 Independently Determine Where Claudins Are Polymerized in Tight-Junction Strand Formation. Cell 2006, 126, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Pulimeno, P.; Paschoud, S.; Citi, S. A role for ZO-1 and PLEKHA7 in recruiting paracingulin to tight and adherens junctions of epithelial cells. J. Biol. Chem. 2011, 286, 16743–16750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Gawlak, G.; Tian, X.; Shah, A.S.; Sarich, N.; Citi, S.; Birukova, A.A. Cingulin role in agonist-induced vascular endothelial permeability. J. Biol. Chem. 2016, 291, 23681–23692. [Google Scholar] [CrossRef] [Green Version]
- Schossleitner, K.; Rauscher, S.; Groger, M.; Friedl, H.P.; Finsterwalder, R.; Habertheuer, A.; Sibilia, M.; Brostjan, C.; Fodinger, D.; Citi, S.; et al. Evidence That Cingulin Regulates Endothelial Barrier Function In Vitro and In Vivo. Arter. Thromb. Vasc. Biol. 2016, 36, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Holzner, S.; Bromberger, S.; Wenzina, J.; Neumuller, K.; Holper, T.M.; Petzelbauer, P.; Bauer, W.; Weber, B.; Schossleitner, K. Phosphorylated cingulin localises GEF-H1 at tight junctions to protect vascular barriers in blood endothelial cells. J. Cell Sci. 2021, 134, jcs258557. [Google Scholar] [CrossRef]
- Guillemot, L.; Citi, S. Cingulin regulates claudin-2 expression and cell proliferation through the small GTPase RhoA. Mol. Biol. Cell 2006, 17, 3569–3577. [Google Scholar] [CrossRef] [Green Version]
- Guillemot, L.; Paschoud, S.; Jond, L.; Foglia, A.; Citi, S. Paracingulin Regulates the Activity of Rac1 and RhoA GTPases by Recruiting Tiam1 and GEF-H1 to Epithelial Junctions. Mol. Biol. Cell 2008, 19, 4442–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemot, L.; Hammar, E.; Kaister, C.; Ritz, J.; Caille, D.; Jond, L.; Bauer, C.; Meda, P.; Citi, S. Disruption of the cingulin gene does not prevent tight junction formation but alters gene expression. J. Cell Sci. 2004, 117, 5245–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemot, L.; Schneider, Y.; Brun, P.; Castagliuolo, I.; Pizzuti, D.; Martines, D.; Jond, L.; Bongiovanni, M.; Citi, S. Cingulin is dispensable for epithelial barrier function and tight junction structure, and plays a role in the control of claudin-2 expression and response to duodenal mucosa injury. J. Cell Sci. 2012, 125, 5005–5012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouaud, F.; Huang, W.; Flinois, A.; Jain, K.; Vasileva, E.; Di Mattia, T.; Mauperin, M.; Parry, D.A.D.; Dugina, V.; Chaponnier, C.; et al. Cingulin and paracingulin tether myosins-2 to junctions to mechanoregulate the plasma membrane. J. Cell Biol. 2023, 322, e202208065. [Google Scholar] [CrossRef]
- Vasileva, E.; Spadaro, D.; Rouaud, F.; King, J.M.; Flinois, A.; Shah, J.; Sluysmans, S.; Mean, I.; Jond, L.; Turner, J.R.; et al. Cingulin binds to the ZU5 domain of scaffolding protein ZO-1 to promote its extended conformation, stabilization, and tight junction accumulation. J. Biol. Chem. 2022, 298, 101797. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Fanning, A.S.; Bridges, A.; Anderson, J.M. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 2009, 20, 3930–3940. [Google Scholar] [CrossRef] [Green Version]
- Paschoud, S.; Jond, L.; Guerrera, D.; Citi, S. PLEKHA7 modulates epithelial tight junction barrier function. Tissue Barriers 2014, 2, e28755. [Google Scholar] [CrossRef] [Green Version]
- Sassi, A.; Wang, Y.; Chassot, A.; Komarynets, O.; Roth, I.; Olivier, V.; Crambert, G.; Dizin, E.; Boscardin, E.; Hummler, E.; et al. Interaction between Epithelial Sodium Channel gamma-Subunit and Claudin-8 Modulates Paracellular Sodium Permeability in Renal Collecting Duct. J. Am. Soc. Nephrol. 2020, 31, 1009–1023. [Google Scholar] [CrossRef]
- Gunzel, D.; Stuiver, M.; Kausalya, P.J.; Haisch, L.; Krug, S.M.; Rosenthal, R.; Meij, I.C.; Hunziker, W.; Fromm, M.; Muller, D. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J. Cell Sci. 2009, 122, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, E.; Sluysmans, S.; Bochaton-Piallat, M.L.; Citi, S. Cell-specific diversity in the expression and organization of cytoplasmic plaque proteins of apical junctions. Ann. N. Y. Acad. Sci. 2017, 1405, 160–166. [Google Scholar] [CrossRef]
- Martinez-Palomo, A.; Meza, I.; Beaty, G.; Cereijido, M. Experimental modulation of occluding junctions in a cultured transporting epithelium. J. Cell Biol. 1980, 87, 736–745. [Google Scholar] [CrossRef]
- Citi, S. Protein kinase inhibitors prevent junction dissociation induced by low extracellular calcium in MDCK epithelial cells. J. Cell Biol. 1992, 117, 169–178. [Google Scholar] [CrossRef]
- Shashikanth, N.; Rizzo, H.E.; Pongkorpsakol, P.; Heneghan, J.F.; Turner, J.R. Electrophysiologic Analysis of Tight Junction Size and Charge Selectivity. Curr. Protoc. 2021, 1, e143. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M. Pores in the wall: Claudins constitute tight junction strands containing aqueous pores. J. Cell Biol. 2000, 149, 13–16. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel Integral Membrane Proteins Localizing at Tight Junctions with No Sequence Similarity to Occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J. Cell Biol. 2001, 153, 263–272. [Google Scholar] [CrossRef]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.; Schulzke, J.D.; Amasheh, S.; Gunzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, S.; Higashi, T.; Furuse, M. ZO-1 Knockout by TALEN-Mediated Gene Targeting in MDCK Cells: Involvement of ZO-1 in the Regulation of Cytoskeleton and Cell Shape. PLoS ONE 2014, 9, e104994. [Google Scholar] [CrossRef] [Green Version]
- Cereijido, M.; Robbins, E.S.; Dolan, W.J.; Rotunno, C.A.; Sabatini, D.D. Polarized monolayers formed by epithelial cells on a permeable and translucent support. J. Cell Biol. 1978, 77, 853–880. [Google Scholar] [CrossRef]
- Paschoud, S.; Citi, S. Inducible overexpression of cingulin in stably transfected MDCK cells does not affect tight junction organization and gene expression. Mol. Membr. Biol. 2008, 25, 1–13. [Google Scholar] [CrossRef]
- Muto, S.; Hata, M.; Taniguchi, J.; Tsuruoka, S.; Moriwaki, K.; Saitou, M.; Furuse, K.; Sasaki, H.; Fujimura, A.; Imai, M.; et al. Claudin-2-deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc. Natl. Acad. Sci. USA 2010, 107, 8011–8016. [Google Scholar] [CrossRef]
- Tamura, A.; Tsukita, S. Paracellular barrier and channel functions of TJ claudins in organizing biological systems: Advances in the field of barriology revealed in knockout mice. Semin. Cell Dev. Biol. 2014, 36, 177–185. [Google Scholar] [CrossRef]
- Flinois, A.; Mean, I.; Mutero-Maeda, A.; Guillemot, L.; Citi, S. Paracingulin recruits CAMSAP3 to tight junctions and regulates microtubule and polarized epithelial cell organization. J. Cell Sci. 2024, 137, jcs260745. [Google Scholar] [CrossRef]
- Sluysmans, S.; Mean, I.; Xiao, T.; Boukhatemi, A.; Ferreira, F.; Jond, L.; Mutero, A.; Chang, C.J.; Citi, S. PLEKHA5, PLEKHA6 and PLEKHA7 bind to PDZD11 to target the Menkes ATPase ATP7A to the cell periphery and regulate copper homeostasis. Mol. Biol. Cell 2021, 32, ar34. [Google Scholar] [CrossRef]
- Guerrera, D.; Shah, J.; Vasileva, E.; Sluysmans, S.; Mean, I.; Jond, L.; Poser, I.; Mann, M.; Hyman, A.A.; Citi, S. PLEKHA7 Recruits PDZD11 to Adherens Junctions to Stabilize Nectins. J. Biol. Chem. 2016, 291, 11016–11029. [Google Scholar] [CrossRef] [Green Version]
- Spadaro, D.; Le, S.; Laroche, T.; Mean, I.; Jond, L.; Yan, J.; Citi, S. Tension-Dependent Stretching Activates ZO-1 to Control the Junctional Localization of Its Interactors. Curr. Biol. 2017, 27, 3783–3795.e3788. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT vs. CGN-KO 1 | |||
|---|---|---|---|
| Gene | Protein | Fold Change of Reads per Gene (log2) | p-Values (−log10) |
| CLDN1 | Claudin-1 | −0.063447 | 0.591236046 |
| CLDN2 | Claudin-2 | 1.53523337 | 3.28460007 |
| CLDN3 | Claudin-3 | −0.499567 | 2.142506518 |
| CLDN4 | Claudin-4 | −0.1789856 | 1.934767597 |
| CLDN6 | Claudin-6 | 0.46861894 | 1.772412443 |
| CLDN7 | Claudin-7 | 0.02805951 | 0.249124176 |
| CLDN9 | Claudin-9 | 0.99625261 | 1.018853025 |
| CLDN15 | Claudin-15 | 0.38513126 | 0.797467008 |
| CLDN16 | Claudin-16 | −0.5573263 | 3.327300283 |
| CLDN23 | Claudin-23 | −0.6045539 | 1.163347307 |
| CLDN34 | Claudin-34 | −0.253871 | 1.311607839 |
| WT vs. CGNL1-KO 1 | |||
|---|---|---|---|
| Gene | Protein | Fold Change of Reads per Gene (log2) | p-Values (−log10) |
| CLDN1 | Claudin-1 | 0.01875057 | 0.135071785 |
| CLDN2 | Claudin-2 | 0.21523078 | 0.232246767 |
| CLDN3 | Claudin-3 | −0.9139707 | 4.300967584 |
| CLDN4 | Claudin-4 | −0.4097344 | 5.170136642 |
| CLDN6 | Claudin-6 | −0.4098655 | 1.30666783 |
| CLDN7 | Claudin-7 | 0.11067803 | 1.478666107 |
| CLDN9 | Claudin-9 | 0.39999074 | 0.272456048 |
| CLDN15 | Claudin-15 | 0.35576133 | 0.696931579 |
| CLDN16 | Claudin-16 | −0.5752402 | 3.413889287 |
| CLDN23 | Claudin-23 | −0.5657903 | 1.031935294 |
| CLDN34 | Claudin-34 | −0.0403864 | 0.13166079 |
| WT vs. Double-KO 2 | |||
|---|---|---|---|
| Gene | Protein | Fold Change of Reads per Gene (log2) | p-Values (−log10) |
| CLDN1 | Claudin-1 | 0.0446277 | 0.377016368 |
| CLDN2 | Claudin-2 | 0.67800971 | 1.059246572 |
| CLDN3 | Claudin-3 | −0.7014976 | 3.236609447 |
| CLDN4 | Claudin-4 | −0.4432871 | 5.592565053 |
| CLDN6 | Claudin-6 | 0.16348518 | 0.420113214 |
| CLDN7 | Claudin-7 | 0.03413621 | 0.315691802 |
| CLDN9 | Claudin-9 | 0.63773487 | 0.518749023 |
| CLDN15 | Claudin-15 | 0.06764114 | 0.093204682 |
| CLDN16 | Claudin-16 | −0.247682 | 1.166960751 |
| CLDN23 | Claudin-23 | −0.7573273 | 1.517074677 |
| CLDN34 | Claudin-34 | −0.0426028 | 0.14099429 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maupérin, M.; Sassi, A.; Méan, I.; Feraille, E.; Citi, S. Knock Out of CGN and CGNL1 in MDCK Cells Affects Claudin-2 but Has a Minor Impact on Tight Junction Barrier Function. Cells 2023, 12, 2004. https://doi.org/10.3390/cells12152004
Maupérin M, Sassi A, Méan I, Feraille E, Citi S. Knock Out of CGN and CGNL1 in MDCK Cells Affects Claudin-2 but Has a Minor Impact on Tight Junction Barrier Function. Cells. 2023; 12(15):2004. https://doi.org/10.3390/cells12152004
Chicago/Turabian StyleMaupérin, Marine, Ali Sassi, Isabelle Méan, Eric Feraille, and Sandra Citi. 2023. "Knock Out of CGN and CGNL1 in MDCK Cells Affects Claudin-2 but Has a Minor Impact on Tight Junction Barrier Function" Cells 12, no. 15: 2004. https://doi.org/10.3390/cells12152004