
IL-18 Signaling in the Rat Central Amygdala Is Disrupted in a Comorbid Model of Post-Traumatic Stress and Alcohol Use Disorder
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
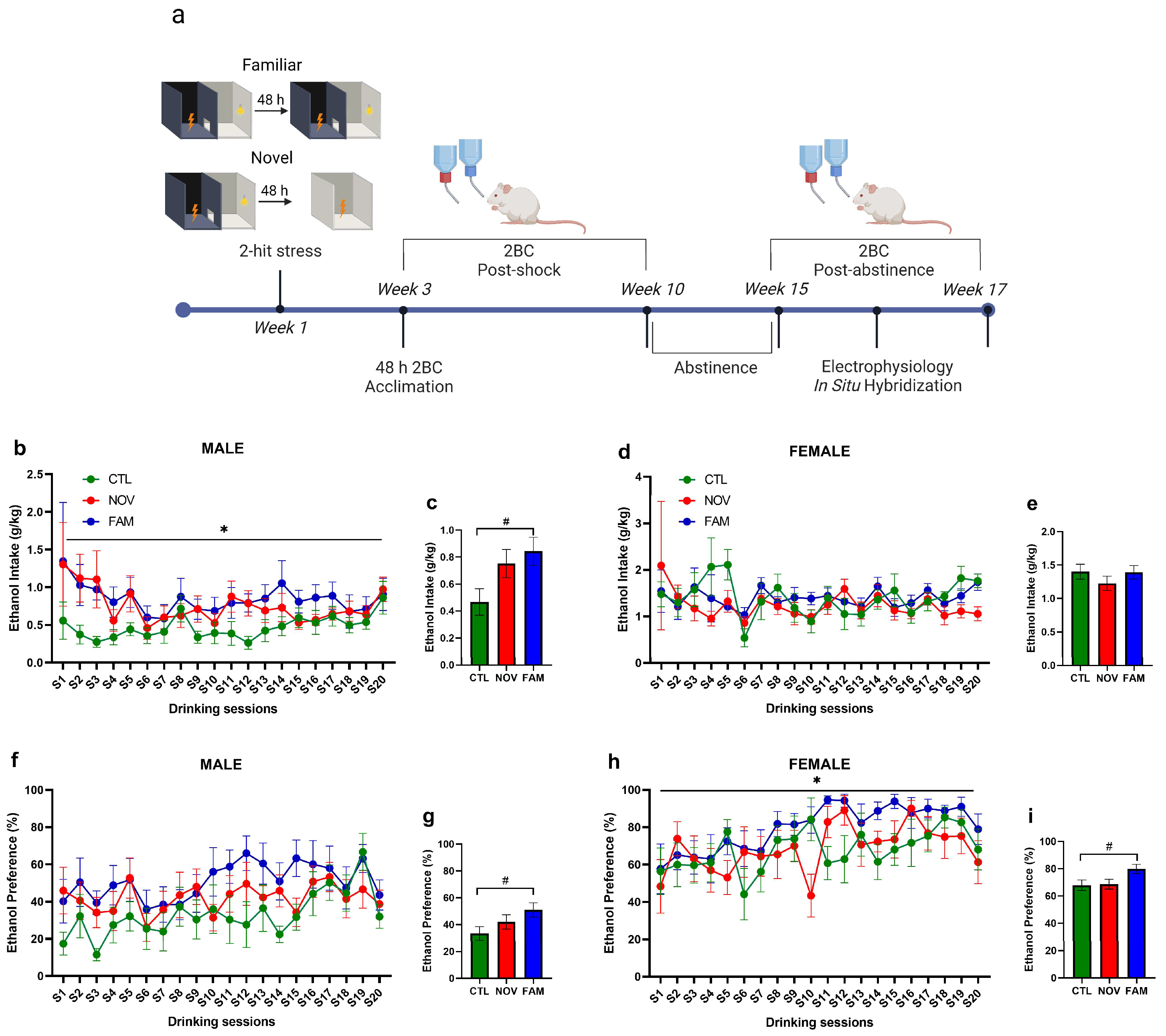
2.2. PTSD/AUD Model Procedures
2.3. RNAscope In Situ Hybridization
2.4. Imaging and Analysis
2.5. Ex Vivo Slice Electrophysiology
2.6. Statistical Analyses
3. Results
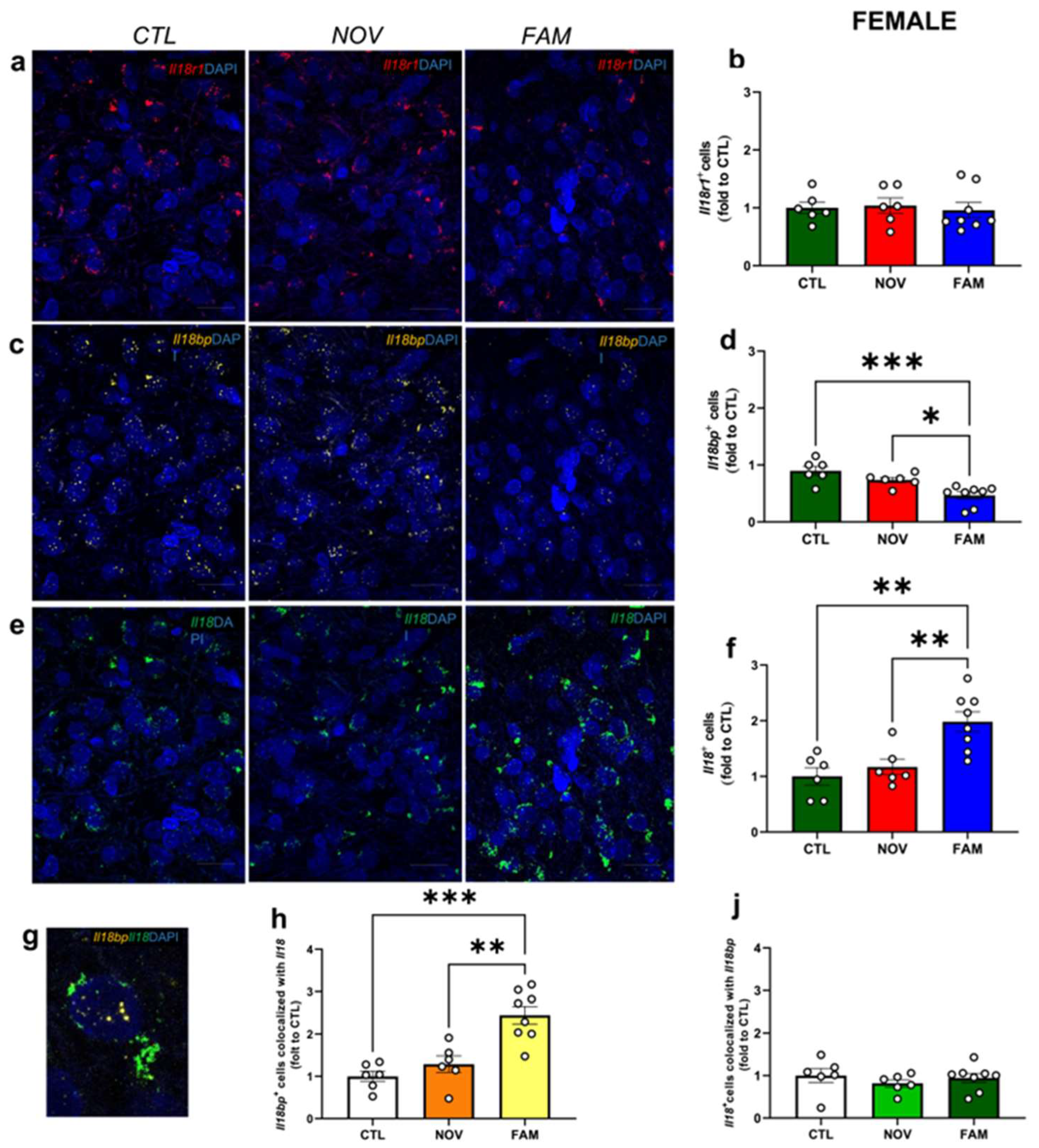
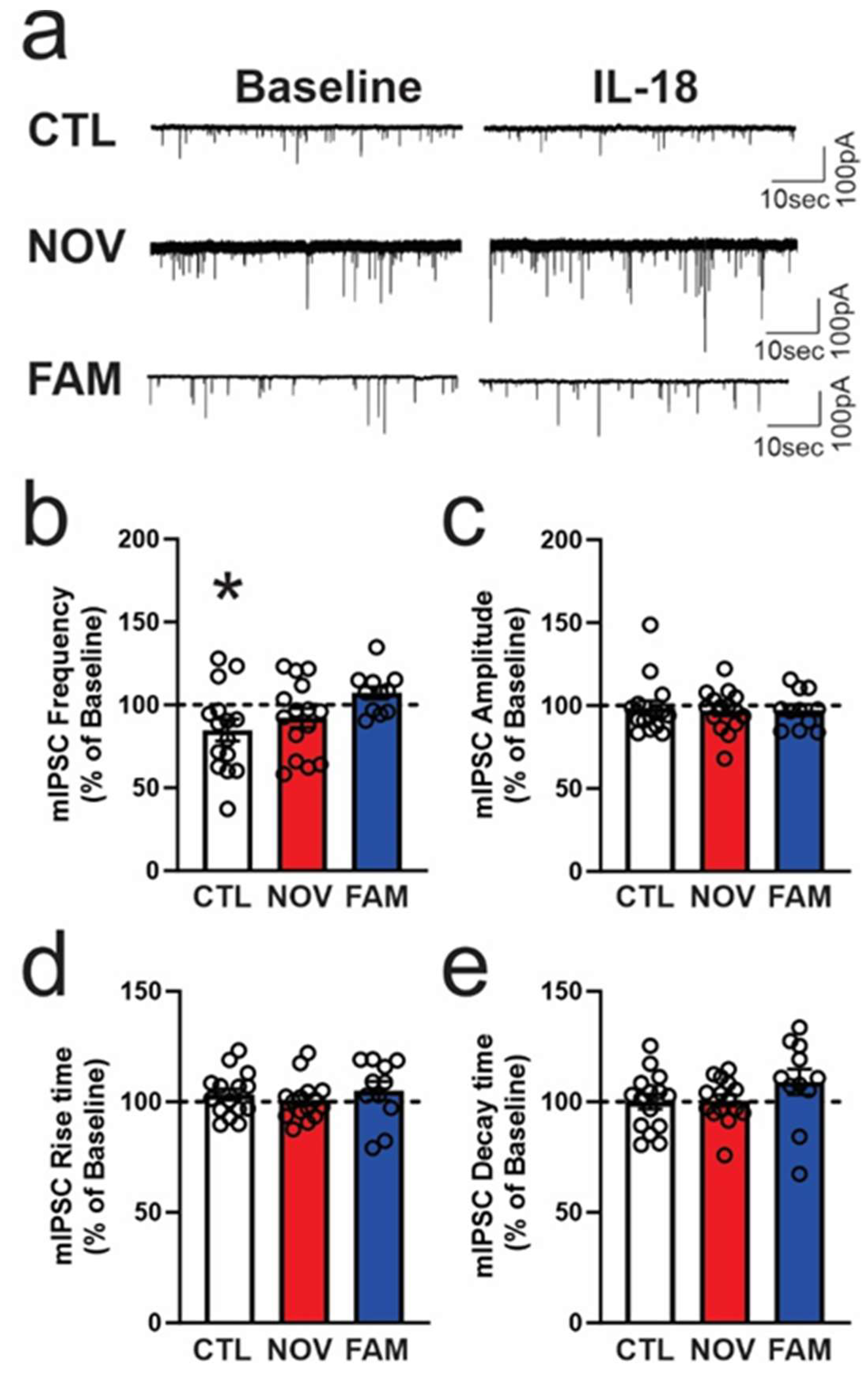
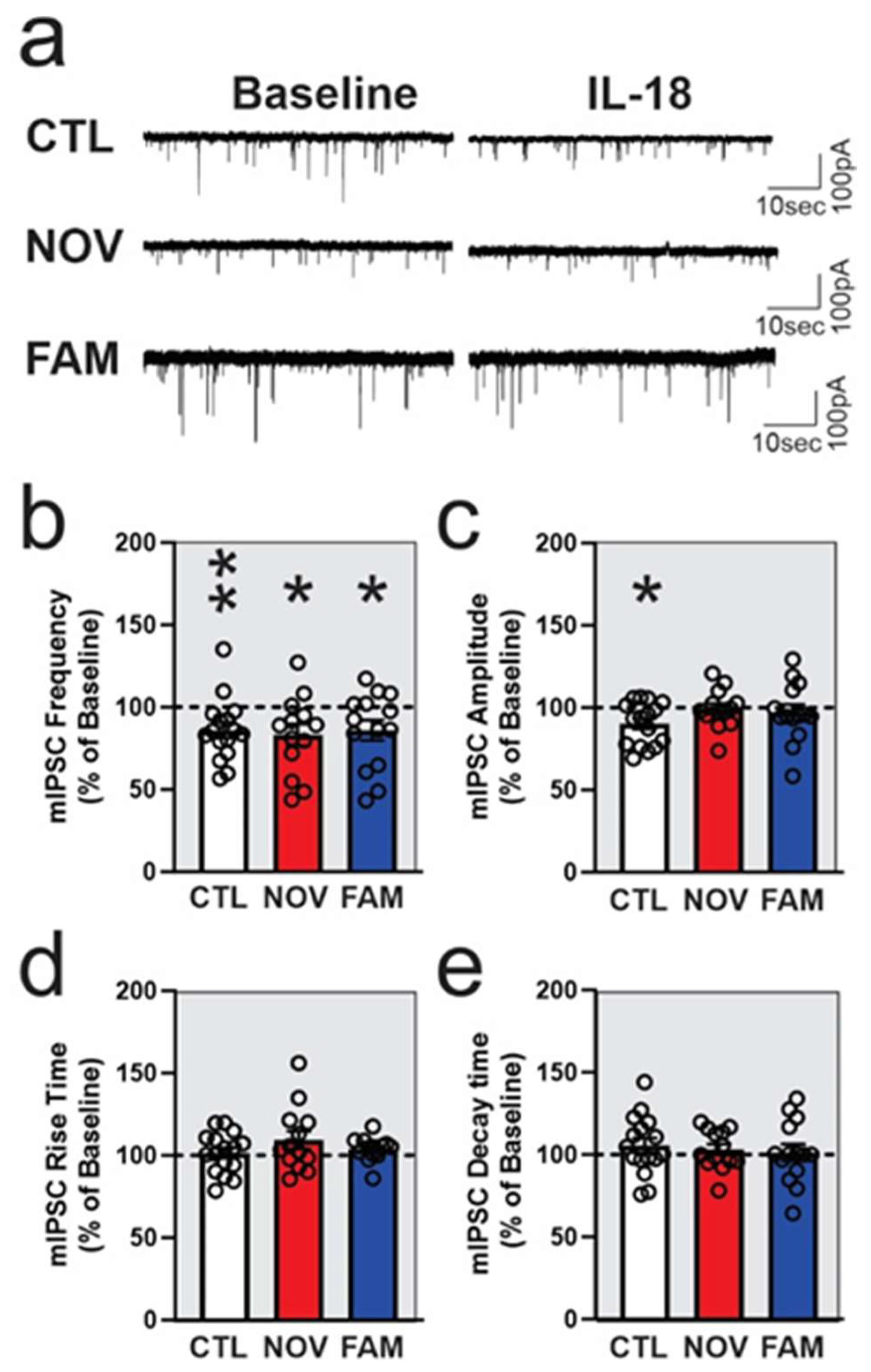
3.1. Expression of Il18r1, Il18 and Il18bp in the CeA of Male and Female PTSD/AUD Rats
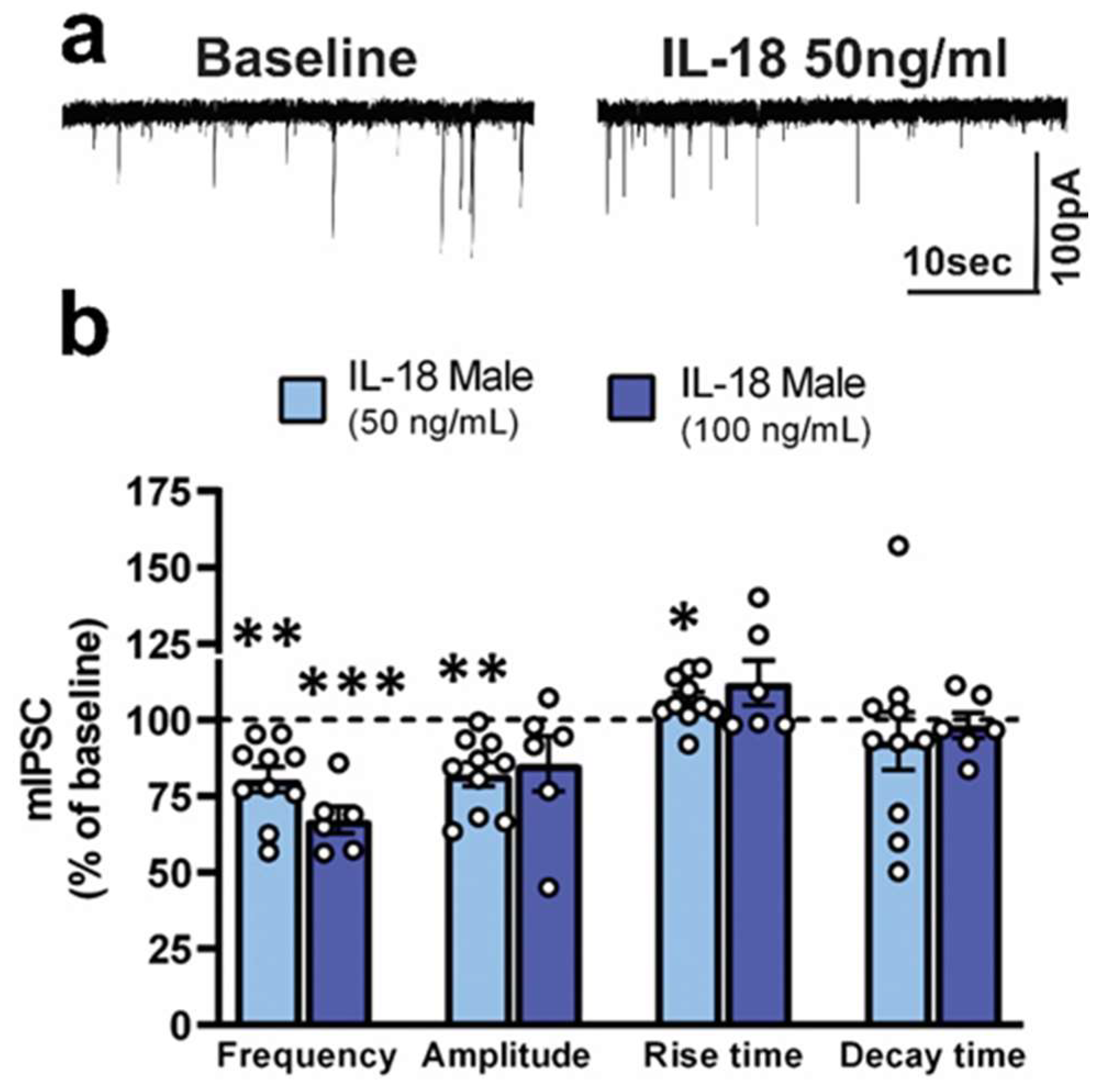
3.2. IL-18 Reduces Vesicular GABA Release in Ex Vivo CeA Slices of Ethanol- and Stress-Naïve Male Rats
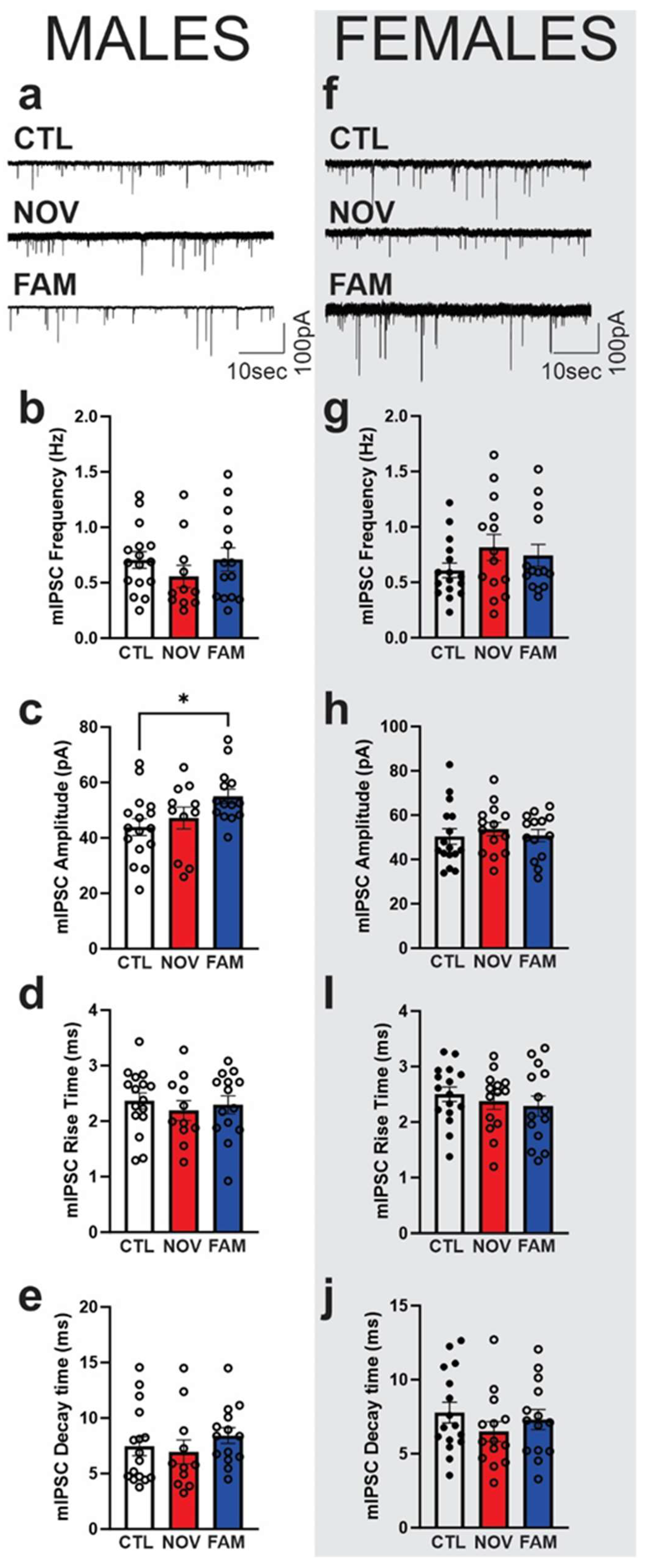
3.3. Stress Alters the Effects of IL-18 on Spontaneous Vesicular GABAergic Transmission in a Sex-Dependent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koob, G.F. Addiction is a Reward Deficit and Stress Surfeit Disorder. Front. Psychiatry 2013, 4, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R. Chronic stress, drug use, and vulnerability to addiction. Ann. N. Y. Acad. Sci. 2008, 1141, 105–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logrip, M.L.; Zorrilla, E.P. Stress history increases alcohol intake in relapse: Relation to phosphodiesterase 10A. Addict. Biol. 2012, 17, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Logrip, M.L.; Zorrilla, E.P.; Koob, G.F. Stress modulation of drug self-administration: Implications for addiction comorbidity with post-traumatic stress disorder. Neuropharmacology 2012, 62, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Johnston, A.L.; Thevos, A.K.; Randall, C.L.; Anton, R.F. Increased severity of alcohol withdrawal in in-patient alcoholics with a co-existing anxiety diagnosis. Br. J. Addict. 1991, 86, 719–725. [Google Scholar] [CrossRef]
- Schellekens, A.F.; de Jong, C.A.; Buitelaar, J.K.; Verkes, R.J. Co-morbid anxiety disorders predict early relapse after inpatient alcohol treatment. Eur. Psychiatry J. Assoc. Eur. Psychiatr. 2015, 30, 128–136. [Google Scholar] [CrossRef]
- Becker, H.C.; Lopez, M.F.; Doremus-Fitzwater, T.L. Effects of stress on alcohol drinking: A review of animal studies. Psychopharmacology 2011, 218, 131–156. [Google Scholar] [CrossRef] [Green Version]
- Gilpin, N.W.; Weiner, J.L. Neurobiology of comorbid post-traumatic stress disorder and alcohol-use disorder. Genes Brain Behav. 2017, 16, 15–43. [Google Scholar] [CrossRef] [Green Version]
- Rorick, L.M.; Finn, P.R.; Steinmetz, J.E. High-alcohol-drinking rats exhibit persistent freezing responses to discrete cues following Pavlovian fear conditioning. Pharmacol. Biochem. Behav. 2003, 76, 223–230. [Google Scholar] [CrossRef]
- Liu, X.; Weiss, F. Stimulus conditioned to foot-shock stress reinstates alcohol-seeking behavior in an animal model of relapse. Psychopharmacology 2003, 168, 184–191. [Google Scholar] [CrossRef]
- Meyer, E.M.; Long, V.; Fanselow, M.S.; Spigelman, I. Stress increases voluntary alcohol intake, but does not alter established drinking habits in a rat model of posttraumatic stress disorder. Alcohol. Clin. Exp. Res. 2013, 37, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Steinman, M.Q.; Kirson, D.; Wolfe, S.A.; Khom, S.; D’Ambrosio, S.R.; Spierling Bagsic, S.R.; Bajo, M.; Vlkolinsky, R.; Hoang, N.K.; Singhal, A.; et al. Importance of sex and trauma context on circulating cytokines and amygdalar GABAergic signaling in a comorbid model of posttraumatic stress and alcohol use disorders. Mol. Psychiatry 2021, 26, 3093–3107. [Google Scholar] [CrossRef]
- Cruz, B.; Vozella, V.; Carper, B.A.; Xu, J.C.; Kirson, D.; Hirsch, S.; Nolen, T.; Bradley, L.; Fain, K.; Crawford, M.; et al. FKBP5 inhibitors modulate alcohol drinking and trauma-related behaviors in a model of comorbid post-traumatic stress and alcohol use disorder. Neuropsychopharmacology 2023, 48, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Bouton, M.E.; Mineka, S.; Barlow, D.H. A modern learning theory perspective on the etiology of panic disorder. Psychol. Rev. 2001, 108, 4–32. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, M.R.; Quinn, J.J.; Fanselow, M.S. The role of muscarinic and nicotinic cholinergic neurotransmission in aversive conditioning: Comparing pavlovian fear conditioning and inhibitory avoidance. Learn. Mem. 2004, 11, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Mayfield, J.; Ferguson, L.; Harris, R.A. Neuroimmune signaling: A key component of alcohol abuse. Curr. Opin. Neurobiol. 2013, 23, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and alcohol. Alcohol. Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef]
- Kimpel, M.W.; Strother, W.N.; McClintick, J.N.; Carr, L.G.; Liang, T.; Edenberg, H.J.; McBride, W.J. Functional gene expression differences between inbred alcohol-preferring and -non-preferring rats in five brain regions. Alcohol 2007, 41, 95–132. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, I.; Wang, S.; Zhang, L.; Harris, R.A.; Mayfield, R.D. Gene co-expression networks in human brain identify epigenetic modifications in alcohol dependence. J. Neurosci. 2012, 32, 1884–1897. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, M.K.; Ponomarev, I.; Hitzemann, R.J.; Belknap, J.K.; Tabakoff, B.; Harris, R.A.; Crabbe, J.C.; Blednov, Y.A.; Grahame, N.J.; Phillips, T.J.; et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc. Natl. Acad. Sci. USA 2006, 103, 6368–6373. [Google Scholar] [CrossRef]
- He, J.; Crews, F.T. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 2008, 210, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Warden, A.S.; Wolfe, S.A.; Khom, S.; Varodayan, F.P.; Patel, R.R.; Steinman, M.Q.; Bajo, M.; Montgomery, S.; Vlkolinsky, R.; Nadav, T.; et al. Microglia control escalation of drinking in alcohol dependent mice: Genomic and synaptic drivers. Biol. Psychiatry 2020, 88, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Nixon, K.; Kim, D.H.; Potts, E.N.; He, J.; Crews, F.T. Distinct cell proliferation events during abstinence after alcohol dependence: Microglia proliferation precedes neurogenesis. Neurobiol. Dis. 2008, 31, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflammation 2010, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Sugama, S.; Conti, B. Interleukin-18 and stress. Brain Res. Rev. 2008, 58, 85–95. [Google Scholar] [CrossRef]
- Kim, T.K.; Kim, J.E.; Choi, J.; Park, J.Y.; Lee, J.E.; Lee, E.H.; Lee, Y.; Kim, B.Y.; Oh, Y.J.; Han, P.L. Local Interleukin-18 System in the Basolateral Amygdala Regulates Susceptibility to Chronic Stress. Mol. Neurobiol. 2017, 54, 5347–5358. [Google Scholar] [CrossRef]
- Kim, T.K.; Kim, J.E.; Choi, J.; Park, J.Y.; Lee, J.E.; Lee, E.H.; Lee, Y.; Kim, B.Y.; Oh, Y.J.; Han, P.L. Erratum to: Local Interleukin-18 System in the Basolateral Amygdala Regulates Susceptibility to Chronic Stress. Mol. Neurobiol. 2017, 54, 5359–5360. [Google Scholar] [CrossRef] [Green Version]
- Lisboa, S.F.; Issy, A.C.; Biojone, C.; Montezuma, K.; Fattori, V.; Del-Bel, E.A.; Guimaraes, F.S.; Cunha, F.Q.; Verri, W.A.; Joca, S.R.L. Mice lacking interleukin-18 gene display behavioral changes in animal models of psychiatric disorders: Possible involvement of immunological mechanisms. J. Neuroimmunol. 2018, 314, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 1999, 10, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Aizawa, Y.; Akita, K.; Taniai, M.; Torigoe, K.; Mori, T.; Nishida, Y.; Ushio, S.; Nukada, Y.; Tanimoto, T.; Ikegami, H.; et al. Cloning and expression of interleukin-18 binding protein. FEBS Lett. 1999, 445, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Blednov, Y.A.; Benavidez, J.M.; Black, M.; Mayfield, J.; Harris, R.A. Role of interleukin-1 receptor signaling in the behavioral effects of ethanol and benzodiazepines. Neuropharmacology 2015, 95, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, M.; Balino, P.; Aragon, C.M.; Guerri, C. Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: Role of TLR4 and TLR2. Neuropharmacology 2015, 89, 352–359. [Google Scholar] [CrossRef]
- Mayeux, J.P.; Teng, S.X.; Katz, P.S.; Gilpin, N.W.; Molina, P.E. Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav. Brain Res. 2015, 279, 22–30. [Google Scholar] [CrossRef]
- Varodayan, F.P.; Pahng, A.R.; Davis, T.D.; Gandhi, P.; Bajo, M.; Steinman, M.Q.; Kiosses, W.B.; Blednov, Y.A.; Burkart, M.D.; Edwards, S.; et al. Chronic ethanol induces a pro-inflammatory switch in interleukin-1beta regulation of GABAergic signaling in the medial prefrontal cortex of male mice. Brain Behav. Immun. 2023, 110, 125–139. [Google Scholar] [CrossRef]
- Patel, R.R.; Khom, S.; Steinman, M.Q.; Varodayan, F.P.; Kiosses, W.B.; Hedges, D.M.; Vlkolinsky, R.; Nadav, T.; Polis, I.; Bajo, M.; et al. IL-1beta expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala. Brain Behav. Immun. 2019, 75, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Novick, D.; Puren, A.J.; Fantuzzi, G.; Shapiro, L.; Muhl, H.; Yoon, D.Y.; Reznikov, L.L.; Kim, S.H.; Rubinstein, M. Overview of interleukin-18: More than an interferon-gamma inducing factor. J. Leukoc. Biol. 1998, 63, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Gracie, J.A.; Forsey, R.J.; Chan, W.L.; Gilmour, A.; Leung, B.P.; Greer, M.R.; Kennedy, K.; Carter, R.; Wei, X.Q.; Xu, D.; et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J. Clin. Investig. 1999, 104, 1393–1401. [Google Scholar] [CrossRef] [Green Version]
- Prossin, A.R.; Koch, A.E.; Campbell, P.L.; McInnis, M.G.; Zalcman, S.S.; Zubieta, J.K. Association of plasma interleukin-18 levels with emotion regulation and mu-opioid neurotransmitter function in major depression and healthy volunteers. Biol. Psychiatry 2011, 69, 808–812. [Google Scholar] [CrossRef]
- Novick, D.; Kim, S.; Kaplanski, G.; Dinarello, C.A. Interleukin-18, more than a Th1 cytokine. Semin. Immunol. 2013, 25, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Ciaramella, A.; Della Vedova, C.; Salani, F.; Viganotti, M.; D’Ippolito, M.; Caltagirone, C.; Formisano, R.; Sabatini, U.; Bossu, P. Increased levels of serum IL-18 are associated with the long-term outcome of severe traumatic brain injury. Neuroimmunomodulation 2014, 21, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hahm, E.; Kim, Y.; Kang, J.; Lee, W.; Han, I.; Myung, P.; Kang, H.; Park, H.; Cho, D. Regulation of IL-18 expression by CRH in mouse microglial cells. Immunol. Lett. 2005, 98, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Han, Q.Q.; Gong, W.Q.; Pan, D.H.; Wang, L.Z.; Hu, W.; Yang, M.; Li, B.; Yu, J.; Liu, Q. Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J. Neuroinflammation 2018, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.X.; Liu, Q.; Cheng, S.; Lei, L.; Lin, A.J.; Wei, R.; Hui, T.C.; Li, Q.; Ao, L.J.; Sham, P.C. Interleukin-18 levels in the hippocampus and behavior of adult rat offspring exposed to prenatal restraint stress during early and late pregnancy. Neural Regen. Res. 2020, 15, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, N.W.; Herman, M.A.; Roberto, M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol. Psychiatry 2015, 77, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Roberto, M.; Kirson, D.; Khom, S. The Role of the Central Amygdala in Alcohol Dependence. Cold Spring Harb. Perspect. Med. 2021, 11, a039339. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.; Shi, C. The extended amygdala: Are the central nucleus of the amygdala and the bed nucleus of the stria terminalis differentially involved in fear versus anxiety? Ann. N. Y. Acad. Sci. 1999, 877, 281–291. [Google Scholar] [CrossRef]
- Janak, P.H.; Tye, K.M. From circuits to behaviour in the amygdala. Nature 2015, 517, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Tasan, R.O.; Bukovac, A.; Peterschmitt, Y.N.; Sartori, S.B.; Landgraf, R.; Singewald, N.; Sperk, G. Altered GABA transmission in a mouse model of increased trait anxiety. Neuroscience 2011, 183, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Khom, S.; Rodriguez, L.; Gandhi, P.; Kirson, D.; Bajo, M.; Oleata, C.S.; Vendruscolo, L.F.; Mason, B.J.; Roberto, M. Alcohol dependence and withdrawal increase sensitivity of central amygdalar GABAergic synapses to the glucocorticoid receptor antagonist mifepristone in male rats. Neurobiol. Dis. 2022, 164, 105610. [Google Scholar] [CrossRef]
- Khom, S.; Steinkellner, T.; Hnasko, T.S.; Roberto, M. Alcohol dependence potentiates substance P/neurokinin-1 receptor signaling in the rat central nucleus of amygdala. Sci. Adv. 2020, 6, eaaz1050. [Google Scholar] [CrossRef] [PubMed]
- Khom, S.; Wolfe, S.A.; Patel, R.R.; Kirson, D.; Hedges, D.M.; Varodayan, F.P.; Bajo, M.; Roberto, M. Alcohol Dependence and Withdrawal Impair Serotonergic Regulation of GABA Transmission in the Rat Central Nucleus of the Amygdala. J. Neurosci. 2020, 40, 6842–6853. [Google Scholar] [CrossRef]
- Roberto, M.; Cruz, M.T.; Gilpin, N.W.; Sabino, V.; Schweitzer, P.; Bajo, M.; Cottone, P.; Madamba, S.G.; Stouffer, D.G.; Zorrilla, E.P.; et al. Corticotropin Releasing Factor-Induced Amygdala Gamma-Aminobutyric Acid Release Plays a Key Role in Alcohol Dependence. Biol. Psychiatry 2010, 67, 831–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberto, M.; Madamba, S.G.; Stouffer, D.G.; Parsons, L.H.; Siggins, G.R. Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci. 2004, 24, 10159–10166. [Google Scholar] [CrossRef] [Green Version]
- Roberto, M.; Madamba, S.G.; Moore, S.D.; Tallent, M.K.; Siggins, G.R. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc. Natl. Acad. Sci. USA 2003, 100, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Varodayan, F.P.; Correia, D.; Kirson, D.; Khom, S.; Oleata, C.S.; Luu, G.; Schweitzer, P.; Roberto, M. CRF modulates glutamate transmission in the central amygdala of naive and ethanol-dependent rats. Neuropharmacology 2017, 125, 418–428. [Google Scholar] [CrossRef]
- Herman, M.A.; Contet, C.; Roberto, M. A Functional Switch in Tonic GABA Currents Alters the Output of Central Amygdala Corticotropin Releasing Factor Receptor-1 Neurons Following Chronic Ethanol Exposure. J. Neurosci. 2016, 36, 10729–10741. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.R.; Wolfe, S.A.; Bajo, M.; Abeynaike, S.; Pahng, A.; Borgonetti, V.; D’Ambrosio, S.; Nikzad, R.; Edwards, S.; Paust, S.; et al. IL-10 normalizes aberrant amygdala GABA transmission and reverses anxiety-like behavior and dependence-induced escalation of alcohol intake. Prog. Neurobiol. 2021, 199, 101952. [Google Scholar] [CrossRef]
- Patel, R.R.; Varodayan, F.P.; Herman, M.A.; Jimenez, V.; Agnore, R.; Gao, L.; Bajo, M.; Cuzon Carlson, V.C.; Walter, N.A.; Fei, S.S.; et al. Synaptic effects of IL-1beta and CRF in the central amygdala after protracted alcohol abstinence in male rhesus macaques. Neuropsychopharmacology 2022, 47, 847–856. [Google Scholar] [CrossRef]
- Jimenez, V.A.; Herman, M.A.; Cuzon Carlson, V.C.; Walter, N.A.; Grant, K.A.; Roberto, M. Synaptic adaptations in the central amygdala and hypothalamic paraventricular nucleus associated with protracted ethanol abstinence in male rhesus monkeys. Neuropsychopharmacology 2019, 44, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Augier, E.; Barbier, E.; Dulman, R.S.; Licheri, V.; Augier, G.; Domi, E.; Barchiesi, R.; Farris, S.; Natt, D.; Mayfield, R.D.; et al. A molecular mechanism for choosing alcohol over an alternative reward. Science 2018, 360, 1321–1326. [Google Scholar] [CrossRef] [Green Version]
- Kirson, D.; Steinman, M.Q.; Wolfe, S.A.; Spierling Bagsic, S.R.; Bajo, M.; Sureshchandra, S.; Oleata, C.S.; Messaoudi, I.; Zorrilla, E.P.; Roberto, M. Sex and context differences in the effects of trauma on comorbid alcohol use and post-traumatic stress phenotypes in actively drinking rats. J. Neurosci. Res. 2021, 99, 3354–3372. [Google Scholar] [CrossRef]
- Wheeler, R.D.; Culhane, A.C.; Hall, M.D.; Pickering-Brown, S.; Rothwell, N.J.; Luheshi, G.N. Detection of the interleukin 18 family in rat brain by RT-PCR. Brain Res. Mol. Brain Res. 2000, 77, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Cervia, D.; Ross, B.; Montanari, C.; Gonzalez, A.S.; Sanchez-Alavez, M.; Marcondes, M.C.; De Vries, D.; Sugama, S.; Brunello, N.; et al. Mapping of the full length and the truncated interleukin-18 receptor alpha in the mouse brain. J. Neuroimmunol. 2009, 214, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alboni, S.; Montanari, C.; Benatti, C.; Blom, J.M.; Simone, M.L.; Brunello, N.; Caggia, F.; Guidotti, G.; Marcondes, M.C.; Sanchez-Alavez, M.; et al. Constitutive and LPS-regulated expression of interleukin-18 receptor beta variants in the mouse brain. Brain Behav. Immun. 2011, 25, 483–493. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tanahashi, T.; Katsuura, S.; Kurokawa, K.; Nishida, K.; Kuwano, Y.; Kawai, T.; Teshima-Kondo, S.; Chikahisa, S.; Tsuruo, Y.; et al. Interleukin-18 deficiency reduces neuropeptide gene expressions in the mouse amygdala related with behavioral change. J. Neuroimmunol. 2010, 229, 129–139. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Sanchez-Alavez, M.; Sugama, S.; Brennan, M.; Fernandez, R.; Bartfai, T.; Conti, B. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 11097–11102. [Google Scholar] [CrossRef]
- Mehta, D.; Gonik, M.; Klengel, T.; Rex-Haffner, M.; Menke, A.; Rubel, J.; Mercer, K.B.; Putz, B.; Bradley, B.; Holsboer, F.; et al. Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: Evidence from endocrine and gene expression studies. Arch. Gen. Psychiatry 2011, 68, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Almli, L.M.; Lori, A.; Meyers, J.L.; Shin, J.; Fani, N.; Maihofer, A.X.; Nievergelt, C.M.; Smith, A.K.; Mercer, K.B.; Kerley, K.; et al. Problematic alcohol use associates with sodium channel and clathrin linker 1 (SCLT1) in trauma-exposed populations. Addict. Biol. 2018, 23, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Swartz, J.R.; Prather, A.A.; Di Iorio, C.R.; Bogdan, R.; Hariri, A.R. A Functional Interleukin-18 Haplotype Predicts Depression and Anxiety through Increased Threat-Related Amygdala Reactivity in Women but Not Men. Neuropsychopharmacology 2017, 42, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Duncan, L.E.; Ratanatharathorn, A.; Aiello, A.E.; Almli, L.M.; Amstadter, A.B.; Ashley-Koch, A.E.; Baker, D.G.; Beckham, J.C.; Bierut, L.J.; Bisson, J.; et al. Largest GWAS of PTSD (N = 20070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol. Psychiatry 2018, 23, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, S.A.; Sidhu, H.; Patel, R.R.; Kreifeldt, M.; D’Ambrosio, S.R.; Contet, C.; Roberto, M. Molecular, Morphological, and Functional Characterization of Corticotropin-Releasing Factor Receptor 1-Expressing Neurons in the Central Nucleus of the Amygdala. eNeuro 2019, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Varodayan, F.P.; Patel, R.R.; Matzeu, A.; Wolfe, S.A.; Curley, D.E.; Khom, S.; Gandhi, P.J.; Rodriguez, L.; Bajo, M.; D’Ambrosio, S.; et al. The Amygdala Noradrenergic System Is Compromised With Alcohol Use Disorder. Biol. Psychiatry 2022, 91, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Kirson, D.; Wolfe, S.A.; Patel, R.R.; Varodayan, F.P.; Snyder, A.E.; Gandhi, P.J.; Khom, S.; Vlkolinsky, R.; Bajo, M.; et al. Alcohol Dependence Induces CRF Sensitivity in Female Central Amygdala GABA Synapses. Int. J. Mol. Sci. 2022, 23, 7842. [Google Scholar] [CrossRef] [PubMed]
- Khom, S.; Borgonetti, V.; Vozella, V.; Kirson, D.; Rodriguez, L.; Gandhi, P.; Bianchi, P.C.; Snyder, A.; Vlkolinsky, R.; Bajo, M.; et al. Glucocorticoid receptors regulate central amygdala GABAergic synapses in Marchigian-Sardinian alcohol-preferring rats. Neurobiol. Stress 2023, 25, 100547. [Google Scholar] [CrossRef]
- Francesconi, W.; Sanchez-Alavez, M.; Berton, F.; Alboni, S.; Benatti, C.; Mori, S.; Nguyen, W.; Zorrilla, E.; Moroncini, G.; Tascedda, F.; et al. The Proinflammatory Cytokine Interleukin 18 Regulates Feeding by Acting on the Bed Nucleus of the Stria Terminalis. J. Neurosci. 2016, 36, 5170–5180. [Google Scholar] [CrossRef] [Green Version]
- Cumiskey, D.; Pickering, M.; O’Connor, J.J. Interleukin-18 mediated inhibition of LTP in the rat dentate gyrus is attenuated in the presence of mGluR antagonists. Neurosci. Lett. 2007, 412, 206–210. [Google Scholar] [CrossRef]
- Curran, B.; O’Connor, J.J. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience 2001, 108, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Nagata, T.; Yamamoto, S.; Okamura, H.; Nishizaki, T. Interleukin-18 stimulates synaptically released glutamate and enhances postsynaptic AMPA receptor responses in the CA1 region of mouse hippocampal slices. Brain Res. 2004, 1012, 190–193. [Google Scholar] [CrossRef]
- Yatsiv, I.; Morganti-Kossmann, M.C.; Perez, D.; Dinarello, C.A.; Novick, D.; Rubinstein, M.; Otto, V.I.; Rancan, M.; Kossmann, T.; Redaelli, C.A.; et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J. Cereb. Blood FlowMetab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2002, 22, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, O.I.; Morganti-Kossmann, M.C.; Heyde, C.E.; Perez, D.; Yatsiv, I.; Shohami, E.; Ertel, W.; Stahel, P.F. Tumor necrosis factor-mediated inhibition of interleukin-18 in the brain: A clinical and experimental study in head-injured patients and in a murine model of closed head injury. J. Neuroinflammation 2004, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, R.; Wheeler, R.D.; Collins, P.D.; Luheshi, G.N.; Pickering-Brown, S.; Kimber, I.; Rothwell, N.J.; Pinteaux, E. Identification of a truncated IL-18R beta mRNA: A putative regulator of IL-18 expressed in rat brain. J. Neuroimmunol. 2003, 145, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Varodayan, F.P.; Soni, N.; Bajo, M.; Luu, G.; Madamba, S.G.; Schweitzer, P.; Parsons, L.H.; Roberto, M. Chronic ethanol exposure decreases CB1 receptor function at GABAergic synapses in the rat central amygdala. Addict. Biol. 2016, 21, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, A.M.; Lowery-Gionta, E.G.; Pleil, K.E.; Li, C.; Sprow, G.M.; Cox, B.R.; Rinker, J.A.; Jijon, A.M.; Pena, J.; Navarro, M.; et al. Central neuropeptide Y modulates binge-like ethanol drinking in C57BL/6J mice via Y1 and Y2 receptors. Neuropsychopharmacology 2012, 37, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Crews, F.T.; Lawrimore, C.J.; Walter, T.J.; Coleman, L.G., Jr. The role of neuroimmune signaling in alcoholism. Neuropharmacology 2017, 122, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Natividad, L.A.; Buczynski, M.W.; Herman, M.A.; Kirson, D.; Oleata, C.S.; Irimia, C.; Polis, I.; Ciccocioppo, R.; Roberto, M.; Parsons, L.H. Constitutive Increases in Amygdalar Corticotropin-Releasing Factor and Fatty Acid Amide Hydrolase Drive an Anxious Phenotype. Biol. Psychiatry 2017, 82, 500–510. [Google Scholar] [CrossRef]
- Takahashi, T.; Iwasaki, A. Sex differences in immune responses. Science 2021, 371, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Turnbull, A.V.; Rivier, C.L. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: Actions and mechanisms of action. Physiol. Rev. 1999, 79, 1–71. [Google Scholar]
- Vecchie, A.; Bonaventura, A.; Toldo, S.; Dagna, L.; Dinarello, C.A.; Abbate, A. IL-18 and infections: Is there a role for targeted therapies? J. Cell Physiol. 2021, 236, 1638–1657. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgonetti, V.; Cruz, B.; Vozella, V.; Khom, S.; Steinman, M.Q.; Bullard, R.; D’Ambrosio, S.; Oleata, C.S.; Vlkolinsky, R.; Bajo, M.; et al. IL-18 Signaling in the Rat Central Amygdala Is Disrupted in a Comorbid Model of Post-Traumatic Stress and Alcohol Use Disorder. Cells 2023, 12, 1943. https://doi.org/10.3390/cells12151943
Borgonetti V, Cruz B, Vozella V, Khom S, Steinman MQ, Bullard R, D’Ambrosio S, Oleata CS, Vlkolinsky R, Bajo M, et al. IL-18 Signaling in the Rat Central Amygdala Is Disrupted in a Comorbid Model of Post-Traumatic Stress and Alcohol Use Disorder. Cells. 2023; 12(15):1943. https://doi.org/10.3390/cells12151943
Chicago/Turabian StyleBorgonetti, Vittoria, Bryan Cruz, Valentina Vozella, Sophia Khom, Michael Q. Steinman, Ryan Bullard, Shannon D’Ambrosio, Christopher S. Oleata, Roman Vlkolinsky, Michal Bajo, and et al. 2023. "IL-18 Signaling in the Rat Central Amygdala Is Disrupted in a Comorbid Model of Post-Traumatic Stress and Alcohol Use Disorder" Cells 12, no. 15: 1943. https://doi.org/10.3390/cells12151943