
Aging-Related Accumulation of Truncated Oxidized Phospholipids Augments Infectious Lung Injury and Endothelial Dysfunction via Cluster of Differentiation 36-Dependent Mechanism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animal Experiments
2.3. Isolation and Culture of Mouse Lung Endothelial Cells
2.4. Precisely Cut Lung Slice Culture
2.5. Real-Time PCR
2.6. Endothelial Permeability
2.7. Statistical Analysis
3. Results
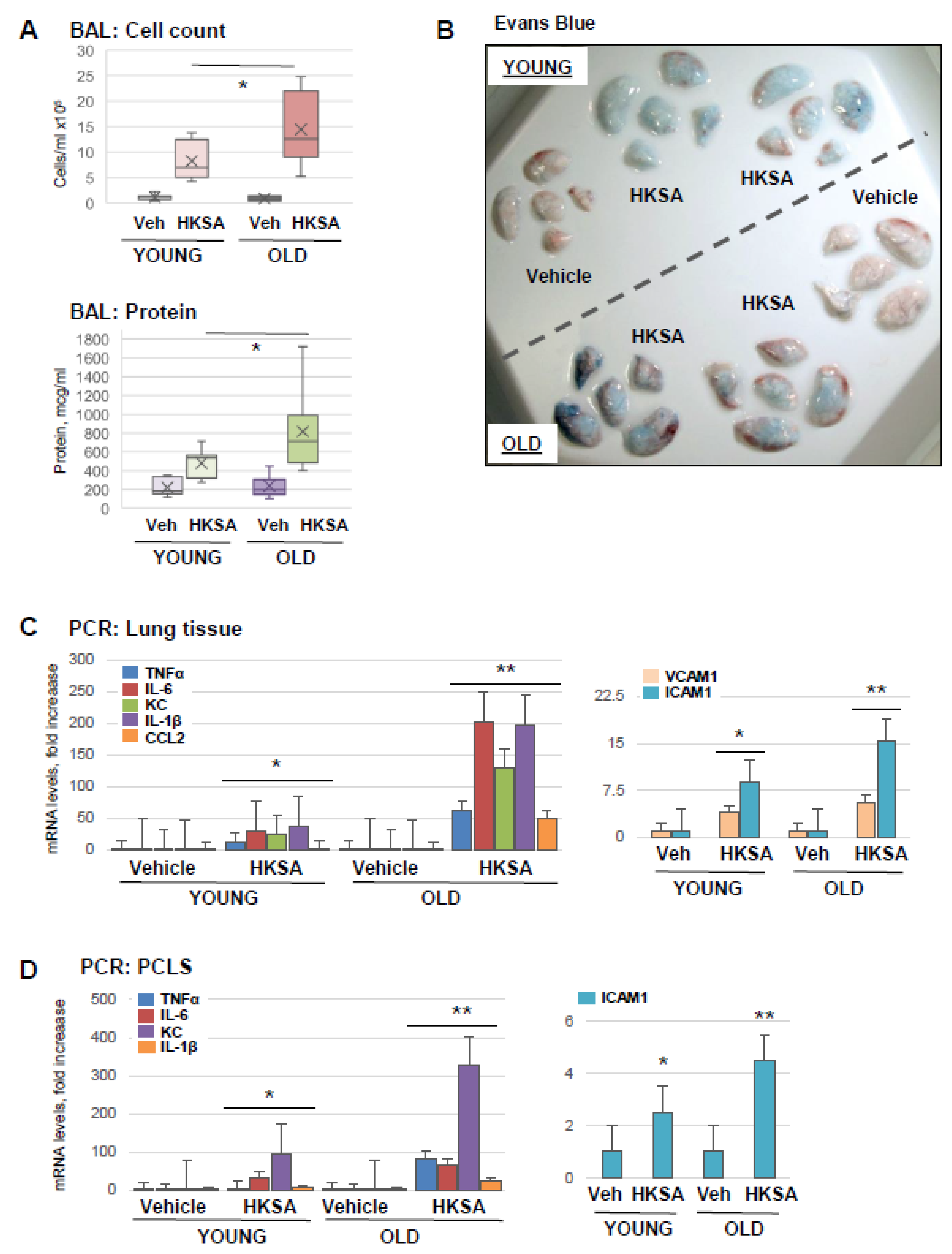
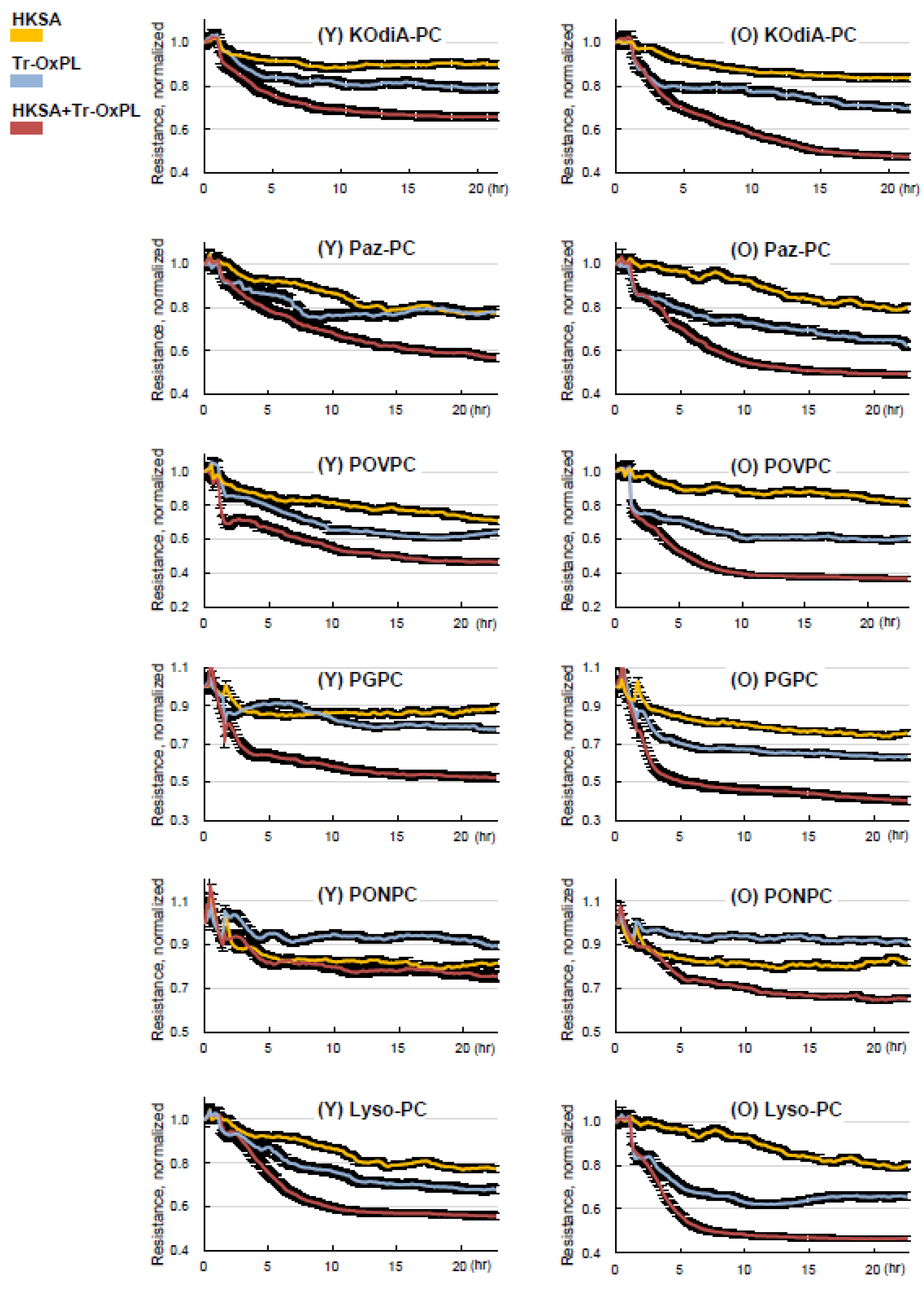
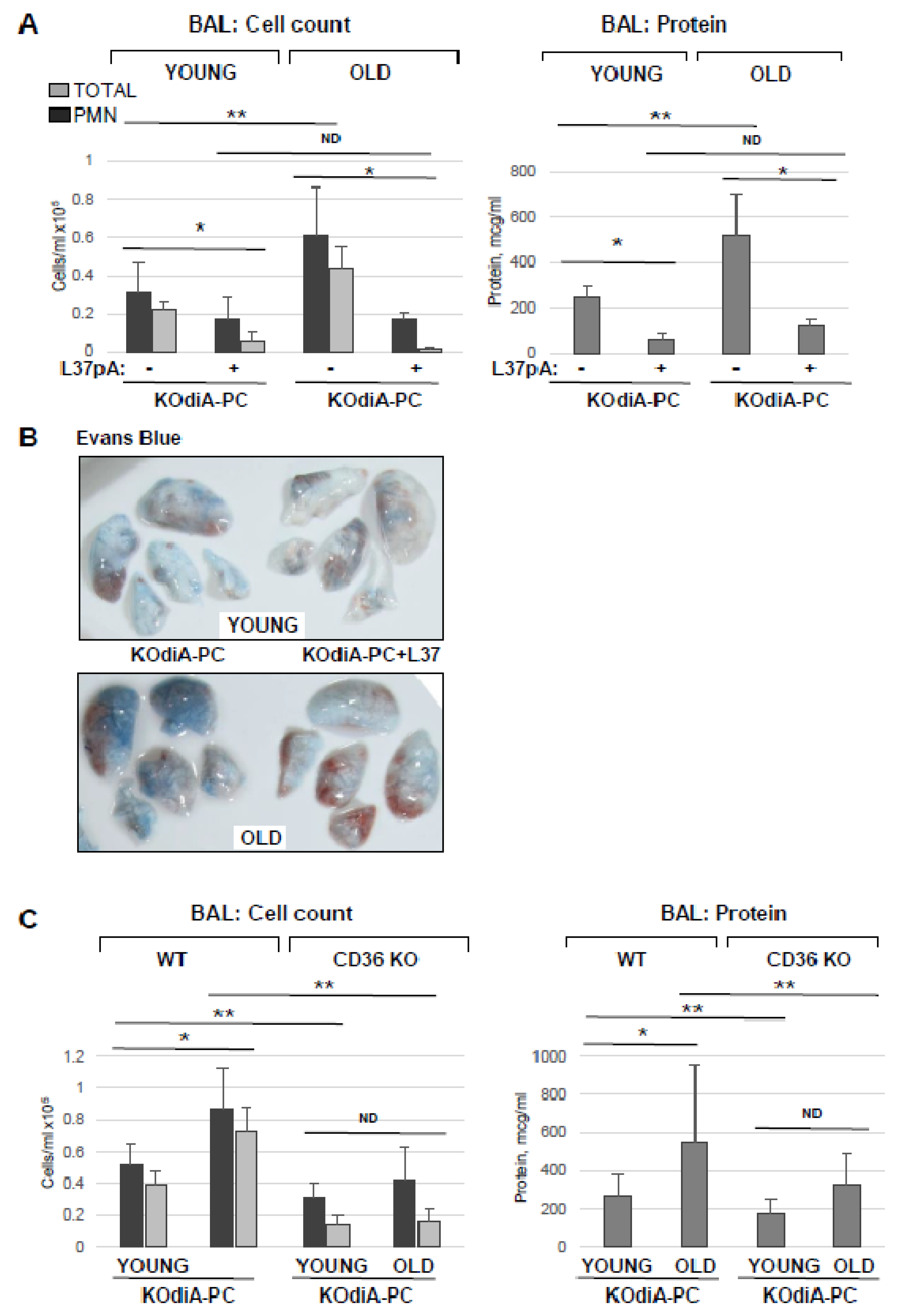
3.1. Old Mice Are More Susceptible to HKSA- and Tr-OxPL-Induced ALI
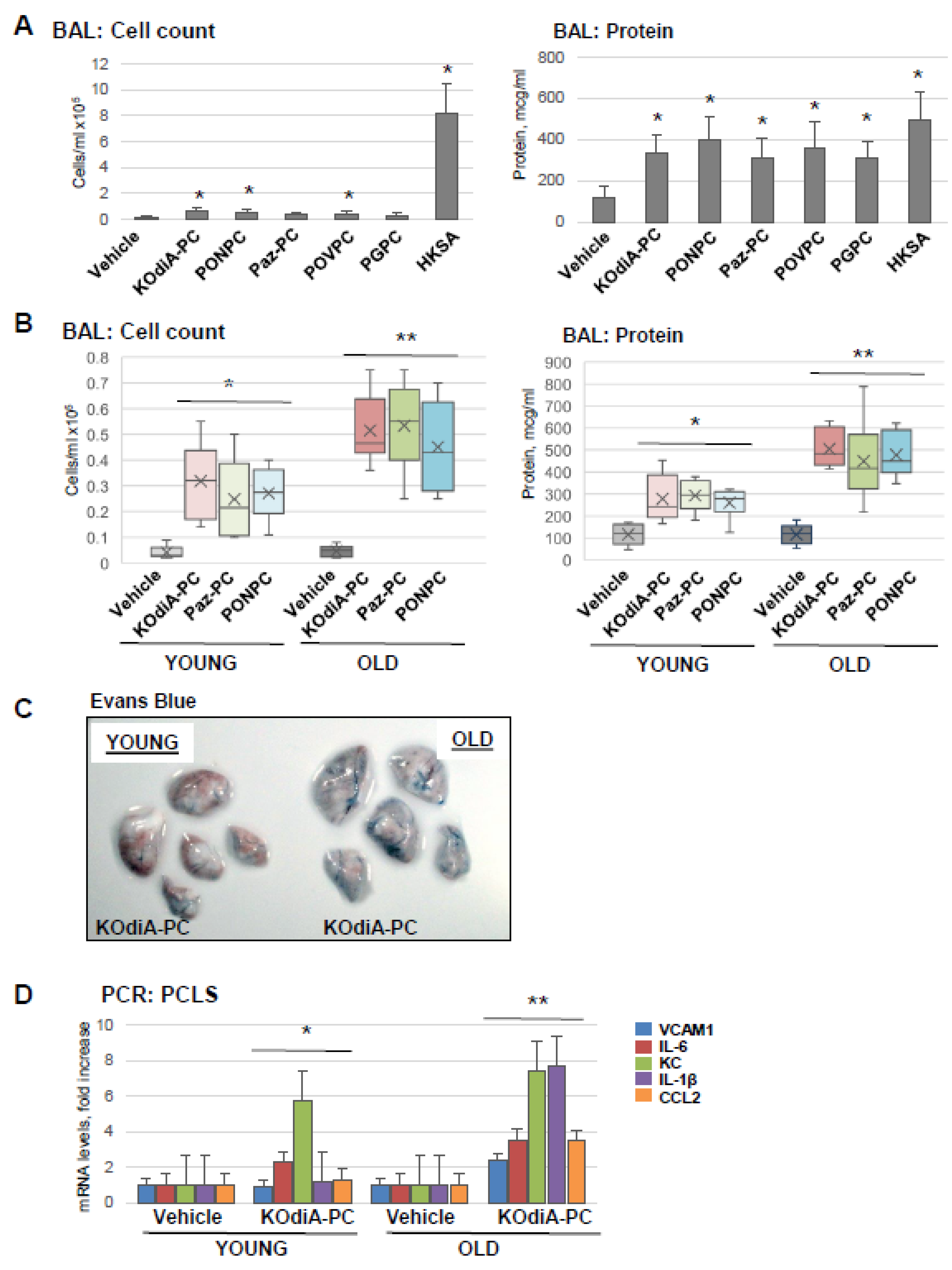
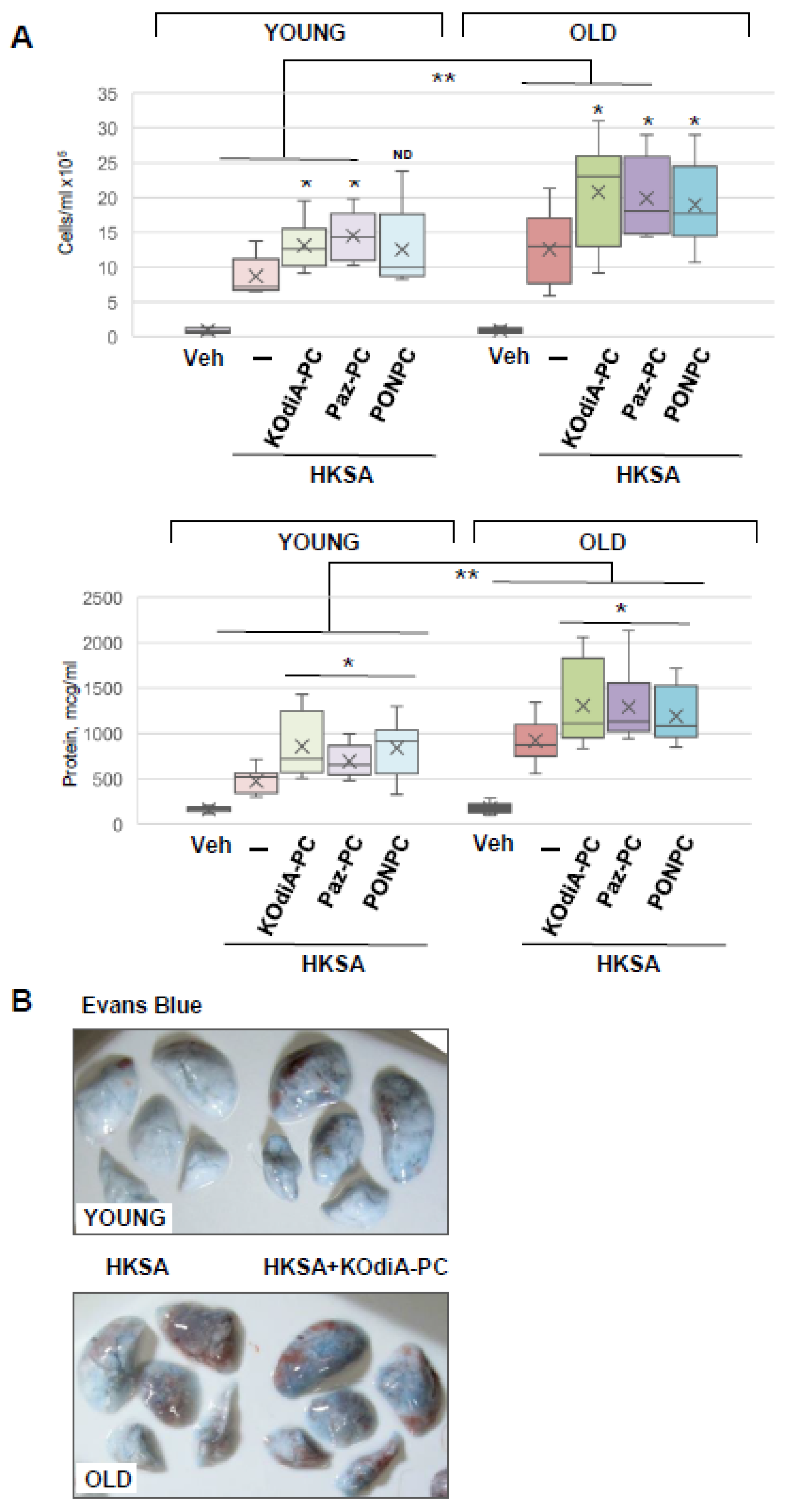
3.2. Tr-OxPLs Exacerbate HKSA-Induced Acute Lung Injury
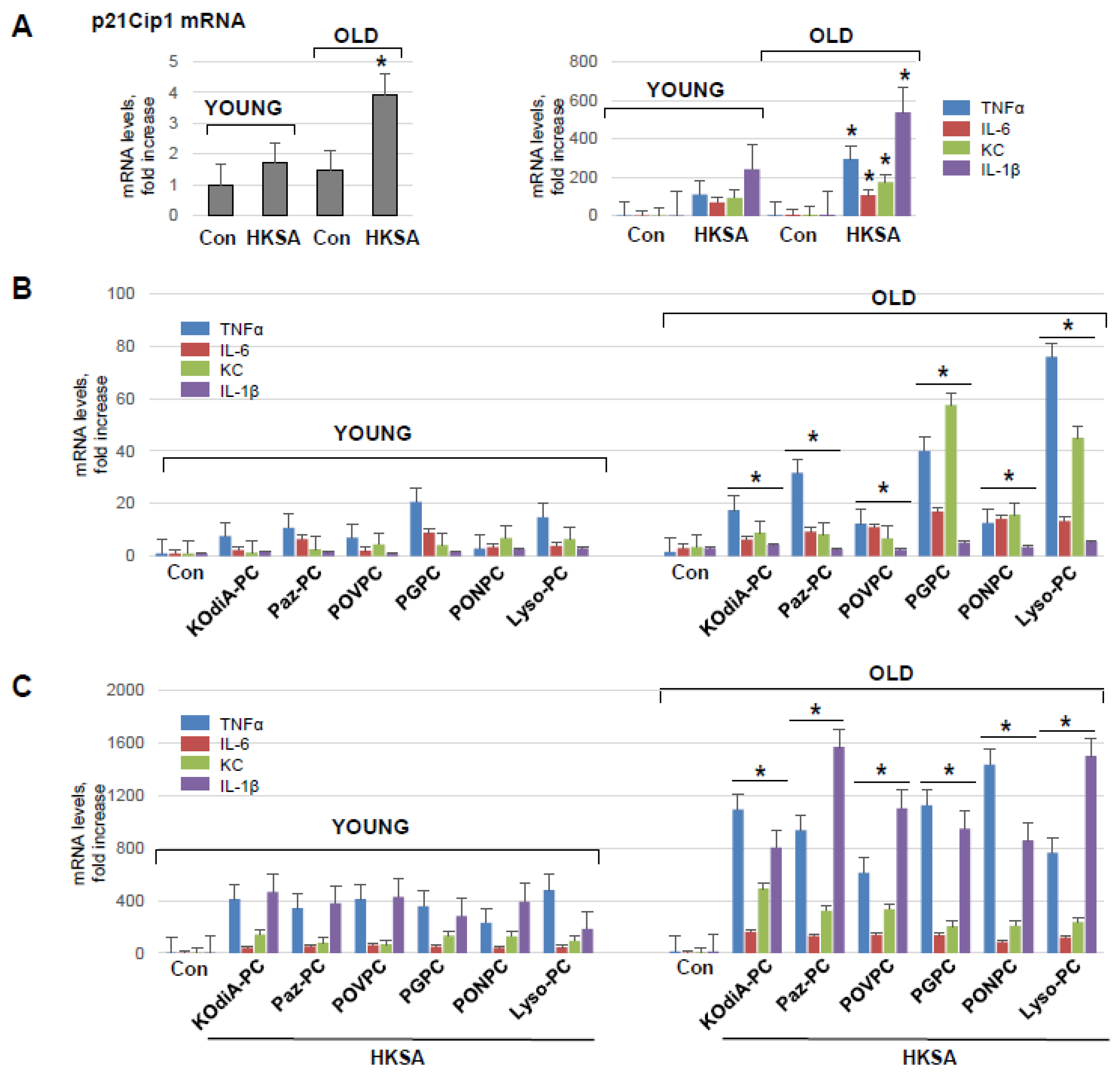
3.3. HKSA and Tr-OxPLs Induce More Severe Inflammation in Endothelial Cell Isolated from Old Mice
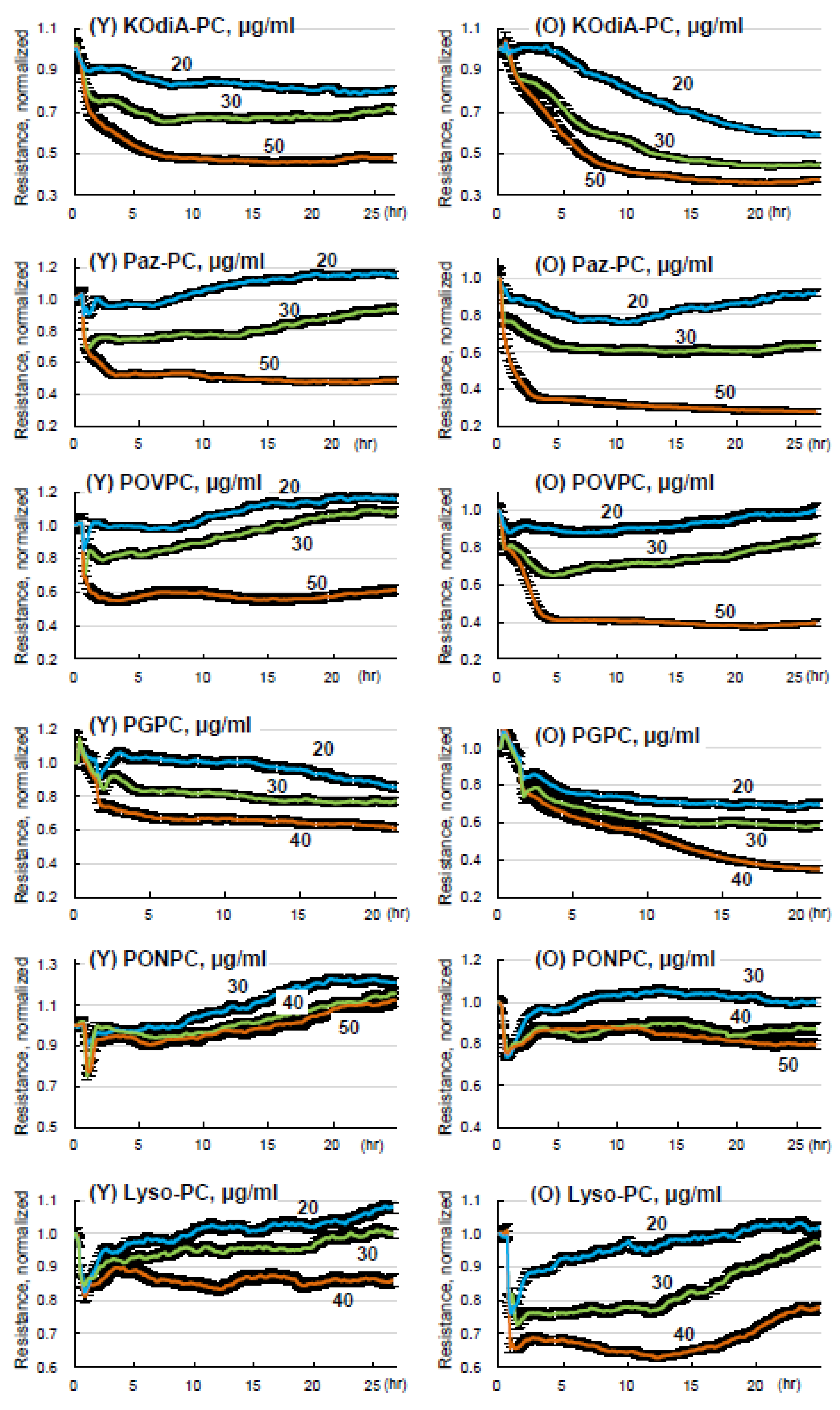
3.4. Lung ECs from Old Mice Are More Susceptible to Tr-OxPLs-Induced Barrier Dysfunction
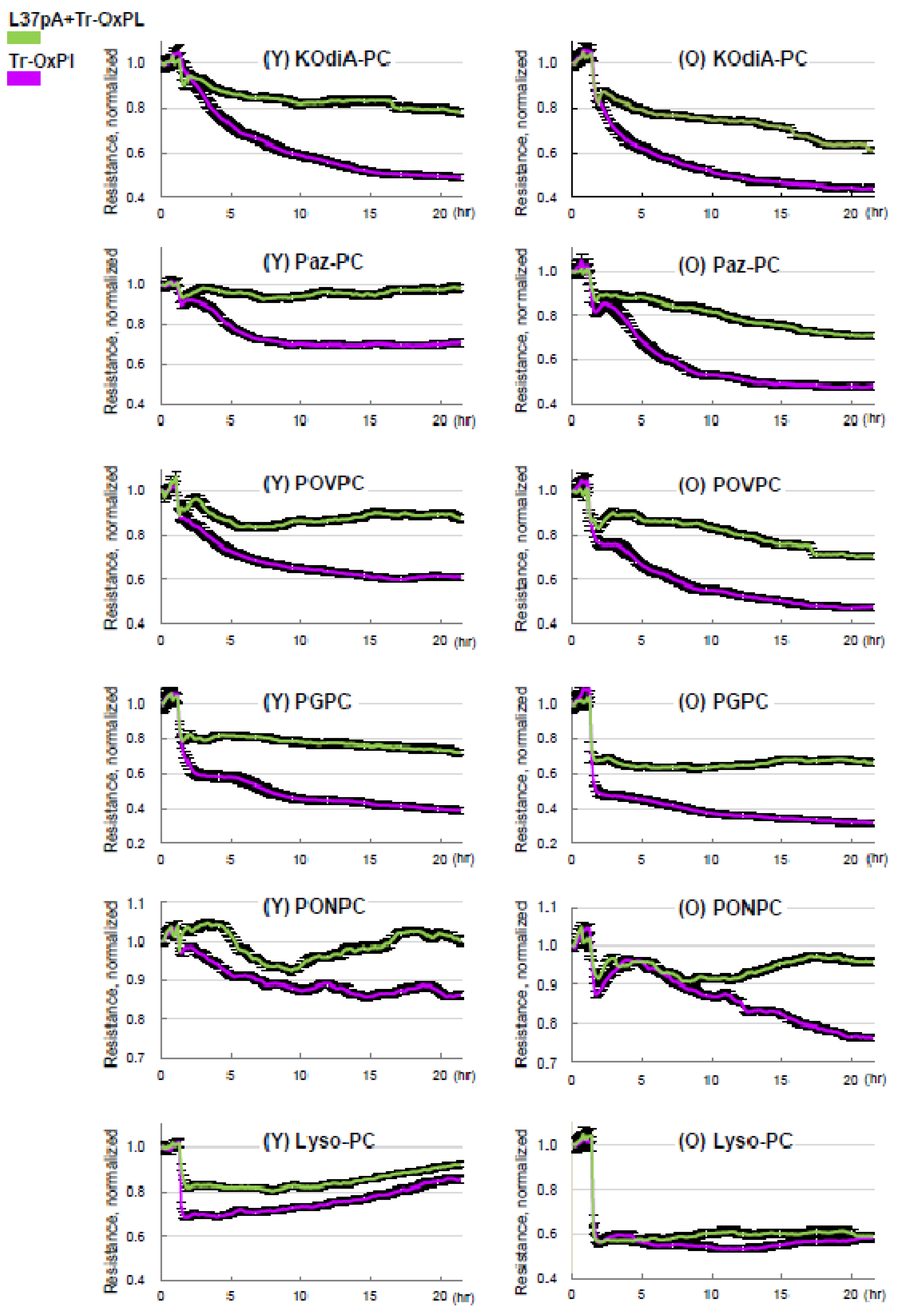
3.5. Inhibition of CD36 Attenuates Tr-OxPLs-Induced ALI Both In Vitro and In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stockl, J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.; Spickett, C.M. Chemistry of phospholipid oxidation. Biochim. Biophys. Acta 2012, 1818, 2374–2387. [Google Scholar] [CrossRef] [Green Version]
- Anthonymuthu, T.S.; Kenny, E.M.; Lamade, A.M.; Kagan, V.E.; Bayir, H. Oxidized phospholipid signaling in traumatic brain injury. Free. Radic. Biol Med. 2018, 124, 493–503. [Google Scholar] [CrossRef]
- Lee, S.; Birukov, K.G.; Romanoski, C.E.; Springstead, J.R.; Lusis, A.J.; Berliner, J.A. Role of phospholipid oxidation products in atherosclerosis. Circ. Res. 2012, 111, 778–799. [Google Scholar] [CrossRef]
- Lichtenstern, C.; Hofer, S.; Mollers, A.; Snyder-Ramos, S.; Spies-Martin, D.; Martin, E.; Schmidt, J.; Motsch, J.; Bardenheuer, H.J.; Weigand, M.A. Lipid peroxidation in acute respiratory distress syndrome and liver failure. J. Surg. Res. 2011, 168, 243–252. [Google Scholar] [CrossRef]
- Wood, L.G.; Gibson, P.G.; Garg, M.L. Biomarkers of lipid peroxidation, airway inflammation and asthma. Eur. Respir. J. 2003, 21, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206.e8. [Google Scholar] [CrossRef]
- Upchurch, C.M.; Yeudall, S.; Pavelec, C.M.; Merk, D.; Greulich, J.; Manjegowda, M.; Raghavan, S.S.; Bochkis, I.M.; Scott, M.M.; Perez-Reyes, E.; et al. Targeting oxidized phospholipids by AAV-based gene therapy in mice with established hepatic steatosis prevents progression to fibrosis. Sci. Adv. 2022, 8, eabn0050. [Google Scholar] [CrossRef]
- Birukova, A.A.; Starosta, V.; Tian, X.; Higginbotham, K.; Koroniak, L.; Berliner, J.A.; Birukov, K.G. Fragmented oxidation products define barrier disruptive endothelial cell response to OxPAPC. Transl. Res. 2013, 161, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Meliton, A.; Shah, A.; Tian, Y.; Ohmura, T.; Sarich, N.; Birukova, A.A.; Birukov, K.G. Role of truncated oxidized phospholipids in acute endothelial barrier dysfunction caused by particulate matter. PLoS ONE 2018, 13, e0206251. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Karki, P.; Kim, J.; Son, S.; Berdyshev, E.; Bochkov, V.N.; Birukova, A.A.; Birukov, K.G. Elevated truncated oxidized phospholipids as a factor exacerbating ALI in the aging lungs. FASEB J. 2019, 33, 3887–3900. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef]
- Martin, G.S.; Mannino, D.M.; Moss, M. The effect of age on the development and outcome of adult sepsis. Crit. Care Med. 2006, 34, 15–21. [Google Scholar] [CrossRef]
- Stengle, J.; Dries, D. Sepsis in the elderly. Crit. Care Nurs. Clin. N. Am. 1994, 6, 421–427. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Liu, J.; Li, W.; Chen, R.; McIntyre, T.M. Circulating biologically active oxidized phospholipids show on-going and increased oxidative stress in older male mice. Redox Biol. 2013, 1, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Frey, B.; Haupt, R.; Alms, S.; Holzmann, G.; Konig, T.; Kern, H.; Kox, W.; Rustow, B.; Schlame, M. Increase in fragmented phosphatidylcholine in blood plasma by oxidative stress. J. Lipid Res. 2000, 41, 1145–1153. [Google Scholar] [CrossRef]
- Kawanishi, N.; Kato, Y.; Yokozeki, K.; Sawada, S.; Sakurai, R.; Fujiwara, Y.; Shinkai, S.; Goda, N.; Suzuki, K. Effects of aging on serum levels of lipid molecular species as determined by lipidomics analysis in Japanese men and women. Lipids Health Dis. 2018, 17, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [CrossRef] [PubMed]
- Boullier, A.; Gillotte, K.L.; Horkko, S.; Green, S.R.; Friedman, P.; Dennis, E.A.; Witztum, J.L.; Steinberg, D.; Quehenberger, O. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J. Biol. Chem. 2000, 275, 9163–9169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Febbraio, M.; Hajjar, D.P.; et al. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 2002, 277, 38517–38523. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Yao, Y.; Huang, C.; Xu, H.; Zhao, Y.; Wang, Y.; Zhu, Y.; Miao, Y.; Feng, X.; Gao, X.; et al. CD36 regulates LPS-induced acute lung injury by promoting macrophages M1 polarization. Cell. Immunol. 2022, 372, 104475. [Google Scholar] [CrossRef]
- Wang, J.; Niu, N.; Xu, S.; Jin, Z.G. A simple protocol for isolating mouse lung endothelial cells. Sci. Rep. 2019, 9, 1458. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Betts, C.; Cunoosamy, D.M.; Aberg, P.M.; Hornberg, J.J.; Sivars, K.B.; Cohen, T.S. Use of precision cut lung slices as a translational model for the study of lung biology. Respir. Res. 2019, 20, 162. [Google Scholar] [CrossRef] [Green Version]
- Pieretti, A.C.; Ahmed, A.M.; Roberts, J.D., Jr.; Kelleher, C.M. A novel in vitro model to study alveologenesis. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Smurova, K.; Birukov, K.G.; Kaibuchi, K.; Garcia, J.G.; Verin, A.D. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc. Res. 2004, 67, 64–77. [Google Scholar] [CrossRef]
- Brown, R.; McKelvey, M.C.; Ryan, S.; Creane, S.; Linden, D.; Kidney, J.C.; McAuley, D.F.; Taggart, C.C.; Weldon, S. The Impact of Aging in Acute Respiratory Distress Syndrome: A Clinical and Mechanistic Overview. Front. Med. 2020, 7, 589553. [Google Scholar] [CrossRef]
- Starosta, V.; Wu, T.; Zimman, A.; Pham, D.; Tian, X.; Oskolkova, O.; Bochkov, V.; Berliner, J.A.; Birukova, A.A.; Birukov, K.G. Differential regulation of endothelial cell permeability by high and low doses of oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine. Am. J. Respir. Cell. Mol. Biol. 2012, 46, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocharov, A.V.; Wu, T.; Baranova, I.N.; Birukova, A.A.; Sviridov, D.; Vishnyakova, T.G.; Remaley, A.T.; Eggerman, T.L.; Patterson, A.P.; Birukov, K.G. Synthetic Amphipathic Helical Peptides Targeting CD36 Attenuate Lipopolysaccharide-Induced Inflammation and Acute Lung Injury. J. Immunol. 2016, 197, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Ke, Y.; Tian, Y.; Ohmura, T.; Sitikov, A.; Sarich, N.; Montgomery, C.P.; Birukova, A.A. Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J. Biol. Chem. 2019, 294, 3369–3384. [Google Scholar] [CrossRef] [Green Version]
- Meliton, A.Y.; Meng, F.; Tian, Y.; Sarich, N.; Mutlu, G.M.; Birukova, A.A.; Birukov, K.G. Oxidized phospholipids protect against lung injury and endothelial barrier dysfunction caused by heat-inactivated Staphylococcus aureus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L550–L562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Letsiou, E.; Wang, H.; Belvitch, P.; Meliton, L.N.; Brown, M.E.; Bandela, M.; Chen, J.; Garcia, J.G.N.; Dudek, S.M. MRSA-induced endothelial permeability and acute lung injury are attenuated by FTY720 S-phosphonate. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L149–L161. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, M.; Abe, A. An Increase of Oxidized Phospholipids and the Role of Macrophages in Intraocular Inflammation. Investig. Ophthalmol. Vis. Sci. 2020, 61, 23. [Google Scholar] [CrossRef]
- Hoffmann, J.; Haendeler, J.; Aicher, A.; Rossig, L.; Vasa, M.; Zeiher, A.M.; Dimmeler, S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 2001, 89, 709–715. [Google Scholar] [CrossRef]
- Brack, C.; Lithgow, G.; Osiewacz, H.; Toussaint, O. EMBO WORKSHOP REPORT: Molecular and cellular gerontology Serpiano, Switzerland, September 18-22, 1999. EMBO J. 2000, 19, 1929–1934. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, Y.; Karki, P.; Li, Y.; Promnares, K.; Zhang, C.-O.; Eggerman, T.L.; Bocharov, A.V.; Birukova, A.A.; Birukov, K.G. Aging-Related Accumulation of Truncated Oxidized Phospholipids Augments Infectious Lung Injury and Endothelial Dysfunction via Cluster of Differentiation 36-Dependent Mechanism. Cells 2023, 12, 1937. https://doi.org/10.3390/cells12151937
Ke Y, Karki P, Li Y, Promnares K, Zhang C-O, Eggerman TL, Bocharov AV, Birukova AA, Birukov KG. Aging-Related Accumulation of Truncated Oxidized Phospholipids Augments Infectious Lung Injury and Endothelial Dysfunction via Cluster of Differentiation 36-Dependent Mechanism. Cells. 2023; 12(15):1937. https://doi.org/10.3390/cells12151937
Chicago/Turabian StyleKe, Yunbo, Pratap Karki, Yue Li, Kamoltip Promnares, Chen-Ou Zhang, Thomas L. Eggerman, Alexander V. Bocharov, Anna A. Birukova, and Konstantin G. Birukov. 2023. "Aging-Related Accumulation of Truncated Oxidized Phospholipids Augments Infectious Lung Injury and Endothelial Dysfunction via Cluster of Differentiation 36-Dependent Mechanism" Cells 12, no. 15: 1937. https://doi.org/10.3390/cells12151937