
PD-L1’s Role in Preventing Alloreactive T Cell Responses Following Hematopoietic and Organ Transplant

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Challenges in Transplantation
2.1. Graft versus Host Disease
2.2. HCT Rejection
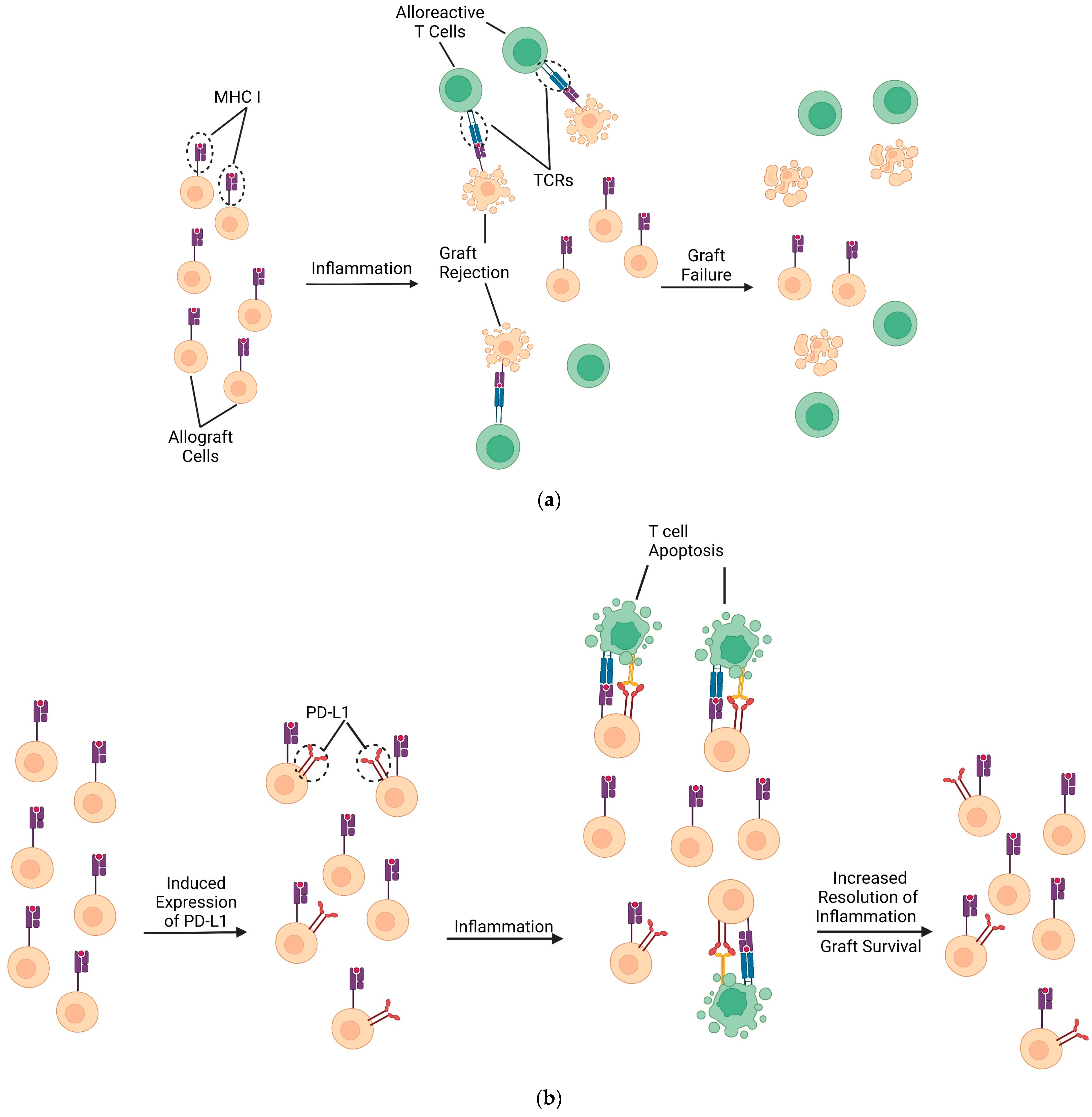
2.3. The Pathophysiologic Process of Transplant Rejection
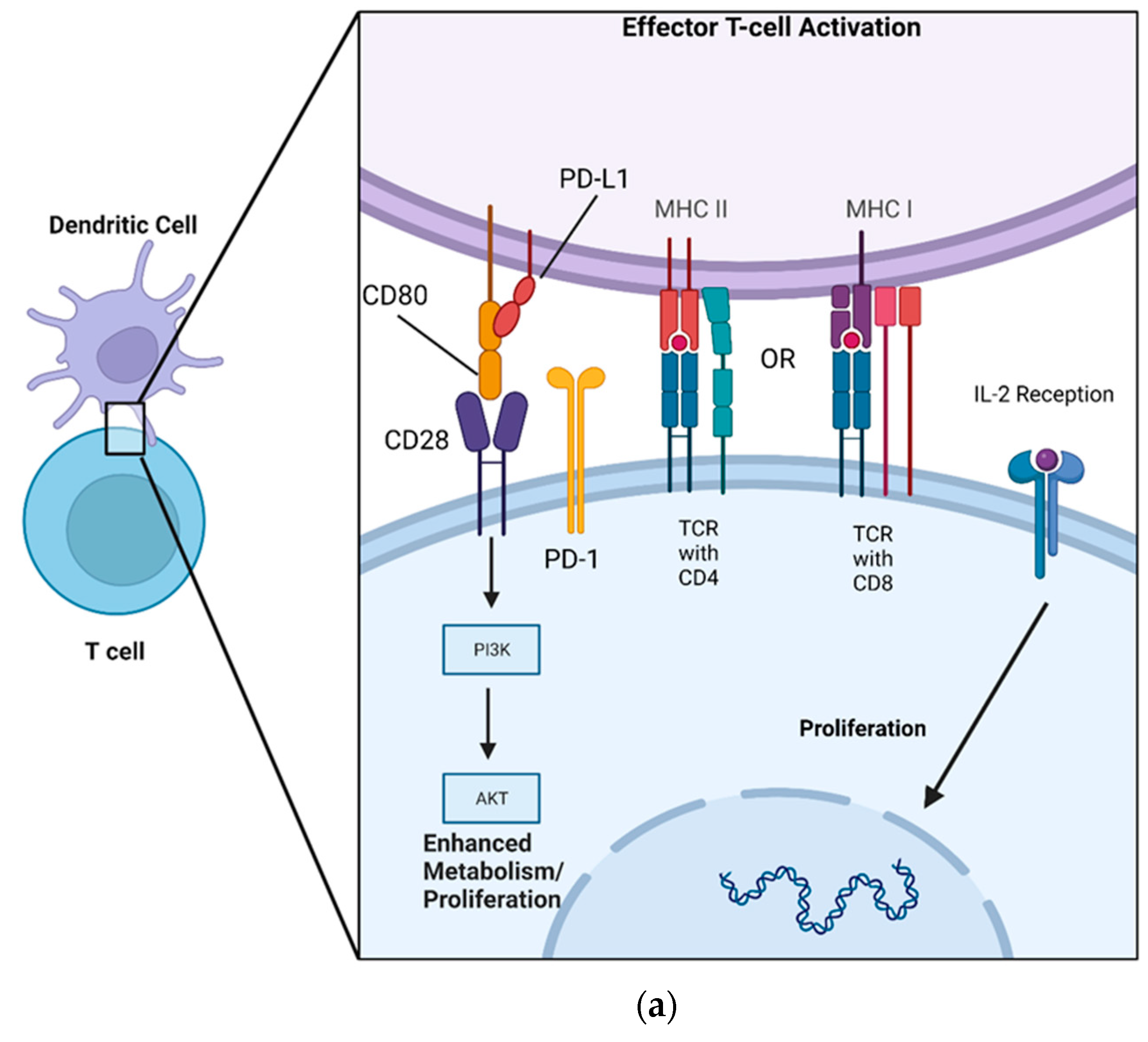
3. PD-L1 Mechanism of Action
4. PD-L1 Amelioration of GVHD, Autoimmunity, and Graft Rejection
4.1. GVHD
4.2. Autoimmunity
4.3. Solid Organ Tolerance

4.4. PD-L1 Viral Reactivation
5. Current State of PD-L1/PD-1 Targeting Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef] [Green Version]
- Felix, N.J.; Allen, P.M. Specificity of T-cell alloreactivity. Nat. Rev. Immunol. 2007, 7, 942–953. [Google Scholar] [CrossRef]
- García, M.A.A.; Yebra, B.G.; Flores, A.L.L.; Guerra, E.G. The major histocompatibility complex in transplantation. J. Transpl. 2012, 2012, 842141. [Google Scholar] [CrossRef]
- Nelson, J.; Alvey, N.; Bowman, L.; Schulte, J.; Segovia, M.C.; McDermott, J.; Te, H.S.; Kapila, N.; Levine, D.J.; Gottlieb, R.L.; et al. Consensus recommendations for use of maintenance immunosuppression in solid organ transplantation: Endorsed by the American College of Clinical Pharmacy, American Society of Transplantation, and International Society for Heart and Lung Transplantation: An executive summary. Pharmacotherapy 2022, 42, 594–598. [Google Scholar] [CrossRef]
- Spierings, E. Minor histocompatibility antigens: Past, present, and future. Tissue Antigens 2014, 84, 360. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Hill, G.R. Alloantigen presentation and graft-versus-host disease: Fuel for the fire. Blood 2016, 127, 2963–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, J.L.M.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Spellman, S.R. Hematology 2022-what is complete HLA match in 2022? Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 83–89. [Google Scholar] [CrossRef]
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell Allorecognition Pathways in Solid Organ Transplantation. Front. Immunol. 2018, 9, 2548. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H. Mechanisms of costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Filipovich, A.H.; Weisdorf, D.; Pavletic, S.; Socie, G.; Wingard, J.R.; Lee, S.J.; Martin, P.; Chien, J.; Przepiorka, D.; Couriel, D.; et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol. Blood Marrow Transpl. 2005, 11, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagasia, M.H.; Greinix, H.T.; Arora, M.; Williams, K.M.; Wolff, D.; Cowen, E.W.; Palmer, J.; Weisdorf, D.; Treister, N.S.; Cheng, G.-S.; et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol. Blood Marrow Transpl. 2015, 21, 389–401.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisdorf, D.; Haake, R.; Blazar, B.; Miller, W.; McGlave, P.; Ramsay, N.; Kersey, J.; Filipovich, A. Treatment of moderate/severe acute graft-versus-host disease after allogeneic bone marrow transplantation: An analysis of clinical risk features and outcome. Blood 1990, 75, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.C.; Young, R.; Devine, S.; Hogan, W.J.; Ayuk, F.; Bunworasate, U.; Chanswangphuwana, C.; Efebera, Y.A.; Holler, E.; Litzow, M.; et al. International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biol. Blood Marrow Transpl. 2016, 22, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiser, R.; Blazar, B.R. Acute Graft-versus-Host Disease—Biologic Process, Prevention, and Therapy. N. Engl. J. Med. 2017, 377, 2167–2179. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Treatment of Pediatric Acute Graft-versus-Host Disease-Lessons from Primary Immunodeficiency? Front. Immunol. 2017, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Alegre, M.L.; Lakkis, F.G.; Morelli, A.E. Antigen Presentation in Transplantation. Trends Immunol. 2016, 37, 831–843. [Google Scholar] [CrossRef]
- Hill, G.R.; Koyama, M. Cytokines and costimulation in acute graft-versus-host disease. Blood 2020, 136, 418–428. [Google Scholar] [CrossRef]
- Xun, C.Q.; Thompson, J.S.; Jennings, C.D.; Brown, S.A.; Widmer, M.B. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood 1994, 83, 2360–2367. [Google Scholar] [CrossRef] [Green Version]
- Cooke, K.R.; Gerbitz, A.; Crawford, J.M.; Teshima, T.; Hill, G.R.; Tesolin, A.; Rossignol, D.P.; Ferrara, J.L. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J. Clin. Investig. 2001, 107, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffner, U.A.; Maeda, Y.; Cooke, K.R.; Reddy, P.; Ordemann, R.; Liu, C.; Ferrara, J.L.M.; Teshima, T. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. J. Immunol. 2004, 172, 7393–7398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, E.K.; Kosaka, H.; Surh, C.D.; Sprent, J.T. Cell contact with Ia antigens on nonhemopoietic cells in vivo can lead to immunity rather than tolerance. J. Exp. Med. 1991, 174, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraverty, R.; Sykes, M. The role of antigen-presenting cells in triggering graft-versus-host disease and graft-versus-leukemia. Blood 2007, 110, 9–17. [Google Scholar] [CrossRef]
- Mohty, M.; Blaise, D.; Faucher, C.; Vey, N.; Bouabdallah, R.; Stoppa, A.-M.; Viret, F.; Gravis, G.; Olive, D.; Gaugler, B. Inflammatory cytokines and acute graft-versus-host disease after reduced-intensity conditioning allogeneic stem cell transplantation. Blood 2005, 106, 4407–4411. [Google Scholar] [CrossRef] [Green Version]
- Nimer, S.D.; Giorgi, J.; Gajewski, J.L.; Ku, N.; Schiller, G.J.; Lee, K.; Territo, M.; Ho, W.; Feio, S.; Selch, M.; et al. Selective depletion of CD8+ cells for prevention of graft-versus-host disease after bone marrow transplantation. A randomized controlled trial. Transplantation 1994, 57, 82–87. [Google Scholar] [CrossRef]
- Via, C.S.; Finkelman, F.D. Critical role of interleukin-2 in the development of acute graft-versus-host disease. Int. Immunol. 1993, 5, 565–572. [Google Scholar] [CrossRef]
- Shustov, A.; Nguyen, P.; Finkelman, F.; Elkon, K.B.; Via, C.S. Related Content Fas Expression on Antigen-Speciic T Cells Has Costimulatory, Helper, and Down-Regulatory Functions In Vivo for Cytotoxic T Cell Responses but Not for T Cell-Dependent B Cell Responses. J. Immunol. 1998, 161, 2848–2855. [Google Scholar] [CrossRef]
- Koyama, M.; Cheong, M.; Markey, K.A.; Gartlan, K.; Kuns, R.D.; Locke, K.R.; Lineburg, K.E.; Teal, B.E.; Mouttie, L.L.-E.; Bunting, M.D.; et al. Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. J. Exp. Med. 2015, 212, 1303–1321. [Google Scholar] [CrossRef] [Green Version]
- Ghayur, T.; Seemayer, T.A.; Kongshavn, P.A.; Gartner, J.G.; Lapp, W.S. Graft-versus-host reactions in the beige mouse. An investigation of the role of host and donor natural killer cells in the pathogenesis of graft-versus-host disease. Transplantation 1987, 44, 261–267. [Google Scholar] [CrossRef]
- Ruggeri, L.; Aversa, F.; Martelli, M.F.; Velardi, A. Allogeneic hematopoietic transplantation and natural killer cell recognition of missing self. Immunol. Rev. 2006, 214, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.M.; Egeler, R.M. Acute GvHD: Pathogenesis and classification. Bone Marrow Transplant. 2008, 41, S58–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Arora, M.; Wang, T.; Spellman, S.R.; He, W.; Couriel, D.R.; Urbano-Ispizua, A.; Cutler, C.S.; Bacigalupo, A.A.; Battiwalla, M.; et al. Increasing Incidence of Chronic Graft-versus-Host Disease inAllogeneic Transplantation: A Report from the Center for International Blood and Marrow Transplant Research. Biol. Blood Marrow Transplant. 2015, 21, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, M.E.D.; Parker, P.M.; Johnston, L.J.; Matos, A.V.B.; Storer, B.; Bensinger, W.I.; Storb, R.; Appelbaum, F.R.; Forman, S.J.; Blume, K.G.; et al. Comparison of chronic graft-versus-host disease after transplantation of peripheral blood stem cells versus bone marrow in allogeneic recipients: Long-term follow-up of a randomized trial. Blood 2002, 100, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Cooke, K.R.; Luznik, L.; Sarantopoulos, S.; Hakim, F.T.; Jagasia, M.; Fowler, D.H.; Brink, M.R.V.D.; Hansen, J.A.; Parkman, R.; Miklos, D.B.; et al. The Biology of Chronic Graft-versus-Host Disease: A Task Force Report from the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease. Biol. Blood Marrow Transplant. 2017, 23, 211–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martires, K.J.; Baird, K.; Citrin, D.E.; Hakim, F.T.; Pavletic, S.Z.; Cowen, E.W. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury: Potential insight into the mechanism of isomorphic and isotopic responses. Arch. Dermatol. 2011, 147, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Nestel, F.P.; Price, K.S.; Seemayer, T.A.; Lapp, W.S. Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor alpha during graft-versus-host disease. J. Exp. Med. 1992, 175, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Rangarajan, H.; Yassai, M.; Subramanian, H.; Komorowski, R.; Whitaker, M.; Gorski, J.; Drobyski, W.R. Emergence of T Cells that recognize nonpolymorphic antigens during graft-versus-host disease. Blood 2012, 119, 6354–6364. [Google Scholar] [CrossRef]
- Van Bekkum, D.W.; Knaan, S. Role of bacterial microflora in development of intestinal lesions from graft-versus-host reaction. J. Natl. Cancer Inst. 1977, 58, 787–790. [Google Scholar] [CrossRef]
- Brüggen, M.C.; Klein, I.; Greinix, H.; Bauer, W.; Kuzmina, Z.; Rabitsch, W.; Kalhs, P.; Petzelbauer, P.; Knobler, R.; Stingl, G.; et al. Diverse T-cell responses characterize the different manifestations of cutaneous graft-versus-host disease. Blood 2014, 123, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Maeda, Y.; Kobayashi, K.; Nishimori, H.; Matsuoka, K.-I.; Fujii, N.; Kondo, E.; Tanaka, T.; Chen, L.; Azuma, M.; et al. Programmed death-1 pathway in host tissues ameliorates Th17/Th1-mediated experimental chronic graft-versus-host disease. J. Immunol. 2014, 193, 2565–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malard, F.; Bossard, C.; Brissot, E.; Chevallier, P.; Guillaume, T.; Delaunay, J.; Mosnier, J.-F.; Moreau, P.; Grégoire, M.; Gaugler, B.; et al. Increased Th17/Treg ratio in chronic liver GVHD. Bone Marrow Transpl. 2014, 49, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, L.; Huang, T.; Geng, S.; Chen, X.; Huang, X.; Lai, P.; Du, X.; Weng, J. The Gut Bacteria Dysbiosis Contributes to Chronic Graft-versus-Host Disease Associated with a Treg/Th1 Ratio Imbalance. Front. Microbiol. 2022, 13, 813576. [Google Scholar] [CrossRef] [PubMed]
- Dudakov, J.A.; Mertelsmann, A.M.; O’connor, M.H.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Boyd, R.L.; Brink, M.R.M.V.D.; Hanash, A.M. Loss of thymic innate lymphoid cells leads to impaired thymopoiesis in experimental graft-versus-host disease. Blood 2017, 130, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Fukushi, N.; Arase, H.; Wang, B.; Ogasawara, K.; Gotohda, T.; Good, R.A.; Onoé, K. Thymus: A direct target tissue in graft-versus-host reaction after allogeneic bone marrow transplantation that results in abrogation of induction of self-tolerance. Proc. Natl. Acad. Sci. USA 1990, 87, 6301–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauri-Hohl, M.M.; Keller, M.P.; Gill, J.; Hafen, K.; Pachlatko, E.; Boulay, T.; Peter, A.; Holländer, G.A.; Krenger, W. Donor T-cell alloreactivity against host thymic epithelium limits T-cell development after bone marrow transplantation. Blood 2007, 109, 4080–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakoda, Y.; Hashimoto, D.; Asakura, S.; Takeuchi, K.; Harada, M.; Tanimoto, M.; Teshima, T. Donor-derived thymic-dependent T Cells cause chronic graft-versus-host disease. Blood 2007, 109, 1756–1764. [Google Scholar] [CrossRef] [Green Version]
- Teshima, T.; Reddy, P.; Liu, C.; Williams, D.; Cooke, K.R.; Ferrara, J.L.M. Impaired thymic negative selection causes autoimmune graft-versus-host disease. Blood 2003, 102, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Young, J.S.; Johnston, H.; Ni, X.; Deng, R.; Racine, J.; Wang, M.; Wang, A.; Todorov, I.; Wang, J.; et al. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T Cells. J. Immunol. 2013, 191, 488–499. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Young, J.S.; Chen, Y.-H.; Shen, E.; Yi, T.; Todorov, I.; Chu, P.G.; Forman, S.J.; Zeng, D. Alloimmune response results in expansion of autoreactive donor CD4+ T Cells in transplants that can mediate chronic graft-versus-host disease. J. Immunol. 2011, 186, 856–868. [Google Scholar] [CrossRef] [Green Version]
- Alho, A.C.; Kim, H.T.; Chammas, M.J.; Reynolds, C.G.; Matos, T.R.; Forcade, E.; Whangbo, J.; Nikiforow, S.; Cutler, C.S.; Koreth, J.; et al. Unbalanced recovery of regulatory and effector T Cells after allogeneic stem cell transplantation contributes to chronic GVHD. Blood 2016, 127, 646–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.L.; Fore, M.S.; Wooten, J.; Roehrs, P.A.; Bhuiya, N.S.; Hoffert, T.; Sharf, A.; Deal, A.M.; Armistead, P.; Coghill, J.; et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood 2012, 120, 2529–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarantopoulos, S.; Stevenson, K.E.; Kim, H.T.; Bhuiya, N.S.; Cutler, C.S.; Soiffer, R.J.; Antin, J.H.; Ritz, J. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin. Cancer Res. 2007, 13, 6107–6114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarantopoulos, S.; Stevenson, K.E.; Kim, H.T.; Cutler, C.S.; Bhuiya, N.S.; Schowalter, M.; Ho, V.T.; Alyea, E.P.; Koreth, J.; Blazar, B.R.; et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood 2009, 113, 3865–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Ma, J.; Peng, R.; Hu, B.; Zhao, Y.; Liu, S.; Hong, J. Biomarkers in Ocular Graft-Versus-Host Disease: Implications for the Involvement of B Cells. Transpl. Cell Ther. 2022, 28, e1–e749. [Google Scholar] [CrossRef]
- Wan, L.; Jin, Z.; Hu, B.; Lv, K.; Lei, L.; Liu, Y.; Song, Y.; Zhu, Y.; Gong, H.; Xu, M.; et al. IL-Y Aggravates Murine Chronic Graft- Versus-Host Disease by Enhancing T and B Cell Responses. Front. Immunol. 2020, 11, 559740. [Google Scholar] [CrossRef]
- Zhai, Z.; Sun, Z.; Li, Q.; Zhang, A.; Liu, H.; Xu, J.; Xu, X.; Geng, L.; Harris, D.; Hu, S.; et al. Correlation of the CD4+CD25high T-regulatory cells in recipients and their corresponding donors to acute GVHD. Transpl. Int. 2007, 20, 440–446. [Google Scholar] [CrossRef]
- Alexander, K.A.; Flynn, R.; Lineburg, K.E.; Kuns, R.D.; Teal, B.E.; Olver, S.D.; Lor, M.; Raffelt, N.C.; Koyama, M.; Leveque, L.; et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J. Clin. Investig. 2014, 124, 4266–4280. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Paz, K.; Flynn, R.; Vulic, A.; Robinson, T.M.; Lineburg, K.E.; Alexander, K.A.; Meng, J.; Roy, S.; Panoskaltsis-Mortari, A.; et al. Pirfenidone ameliorates murine chronic GVHD through inhibition of macrophage infiltration and TGF-β production. Blood 2017, 129, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.; Du, J.; Veenstra, R.G.; Reichenbach, D.K.; Panoskaltsis-Mortari, A.; Taylor, P.A.; Freeman, G.J.; Serody, J.; Murphy, W.J.; Munn, D.; et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood 2014, 123, 3988–3998. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Lai, P.; Wang, Y.; Huang, T.; Chen, X.; Geng, S.; Huang, X.; Luo, C.; Wu, S.; Ling, W.; et al. Extracellular vesicles derived from mesenchymal stem cells prevent skin fibrosis in the cGVHD mouse model by suppressing the activation of macrophages and B cells immune response. Int. Immunopharmacol. 2020, 84, 106541. [Google Scholar] [CrossRef] [PubMed]
- Kuroi, T.; Fujii, N.; Ichimura, K.; Seike, K.; Yamamoto, A.; Kambara, Y.; Sugimoto, S.; Otani, S.; Saeki, K.; Fujiwara, H.; et al. Characterization of localized macrophages in bronchiolitis obliterans after allogeneic hematopoietic cell transplantation. Int. J. Hematol. 2021, 114, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Panoskaltsis-Mortari, A.; Tram, K.V.; Price, A.P.; Wendt, C.H.; Blazar, B.R. A new murine model for bronchiolitis obliterans post-bone marrow transplant. Am. J. Respir. Crit. Care Med. 2007, 176, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Flynn, R.; Price, A.; Ranger, A.; Browning, J.L.; Taylor, P.A.; Ritz, J.; Antin, J.H.; Murphy, W.J.; Luznik, L.; et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood 2012, 119, 1570–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, S.; George, B.; Savani, B.N. Bleeding and thrombotic complications. In The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; Springer: Berlin/Heidelberg, Germany, 2018; pp. 301–306. [Google Scholar] [CrossRef]
- Shatry, A.M.; Roopenian, D.C.; Levy, R.B. Survival and function of MiHA epitope-specific host CD8 TM cells following ablative conditioning and HCT. Biol. Blood Marrow Transpl. 2007, 13, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Marijt, W.A.; Kernan, N.A.; Diaz-Barrientos, T.; Veenhof, W.F.; O’Reilly, R.J.; Willemze, R.; Falkenburg, J.H. Multiple minor histocompatibility antigen-specific cytotoxic T lymphocyte clones can be generated during graft rejection after HLA-identical bone marrow transplantation. Bone Marrow Transpl. 1995, 16, 125–132. [Google Scholar]
- Marijt, W.A.; Veenhof, W.F.; Brand, A.; Goulmy, E.; Fibbe, W.E.; Willemze, R.; van Rood, J.J.; Falkenburg, J.H. Minor histocompatibility antigen-specific cytotoxic T Cell lines, capable of lysing human hematopoietic progenitor cells, can be generated in vitro by stimulation with HLA-identical bone marrow cells. J. Exp. Med. 1991, 173, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Ward, B.; Schatzle, J.D.; Bennett, M. Perforin- and Fas-dependent mechanisms of natural killer cell-mediated rejection of incompatible bone marrow cell grafts. Eur. J. Immunol. 2002, 32, 793–799. [Google Scholar] [CrossRef]
- Bennett, M.; Taylor, P.A.; Austin, M.; Baker, M.B.; Schook, L.; Rutherford, M.S.; Kumar, V.; Podack, E.R.; Mohler, K.M.; Levy, R.B.; et al. Cytokine and cytotoxic pathways of NK cell rejection of class I-deficient bone marrow grafts: Influence of mouse colony environment. Int. Immunol. 1998, 10, 785–790. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, Z.; Shatry, A.; Deyev, V.; Podack, E.; Mammolenti, M.; Blazar, B.R.; Yagita, H.; Levy, R.B. Effector cells derived from host CD8 memory T Cells mediate rapid resistance against minor histocompatibility antigen-mismatched allogeneic marrow grafts without participation of perforin, Fas ligand, and the simultaneous inhibition of 3 tumor necrosis factor family effector pathways. Biol. Blood Marrow Transpl. 2005, 11, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Mammolenti, M.; Jones, M.; Jurecic, R.; Sayers, T.J.; Levy, R.B. Antigen-primed CD8+ T Cells can mediate resistance, preventing allogeneic marrow engraftment in the simultaneous absence of perforin-, CD95L-, TNFR1-, and TRAIL-dependent killing. Blood 2003, 101, 3991–3999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merli, P.; Caruana, I.; De Vito, R.; Strocchio, L.; Weber, G.; Del Bufalo, F.; Buatois, V.; Montanari, P.; Cefalo, M.G.; Pitisci, A.; et al. Role of interferon-γ in immune-mediated graft failure after allogeneic hematopoietic stem cell transplantation. Haematologica 2019, 104, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Hashimoto, D.; Nagafuji, K.; Eto, T.; Ohno, Y.; Aoyama, K.; Iwasaki, H.; Miyamoto, T.; Hill, G.; Akashi, K.; et al. Expansion of donor-reactive host T Cells in primary graft failure after allogeneic hematopoietic SCT following reduced-intensity conditioning. Bone Marrow Transpl. 2014, 49, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Lapidot, T.; Faktorowich, Y.; Lubin, I.; Reisner, Y. Enhancement of T-cell-depleted bone marrow allografts in the absence of graft-versus-host disease is mediated by CD8+ CD4− and not by CD8- CD4+ thymocytes. Blood 1992, 80, 2406–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.J. Prevention of allogeneic marrow graft rejection by donor T Cells that do not recognize recipient alloantigens: Potential role of a veto mechanism. Blood 1996, 88, 962–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.J. Donor CD8 cells prevent allogeneic marrow graft rejection in mice: Potential implications for marrow transplantation in humans. J. Exp. Med. 1993, 178, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, W.J.; Bennett, M.; Kumar, V.; Longo, D.L. Donor-type activated natural killer cells promote marrow engraftment and B cell development during allogeneic bone marrow transplantation. J. Immunol. 1992, 148, 2953–2960. [Google Scholar] [CrossRef]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Celso, C.L.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef] [Green Version]
- Joffre, O.; Santolaria, T.; Calise, D.; Al Saati, T.; Hudrisier, D.; Romagnoli, P.; Van Meerwijk, J.P.M. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 2008, 14, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Barao, I.; Hanash, A.M.; Hallett, W.; Welniak, L.A.; Sun, K.; Redelman, D.; Blazar, B.R.; Levy, R.B.; Murphy, W.J. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 5460–5465. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Lee, J.-H.; Lee, J.-H.; Park, H.-S.; Choi, E.-J.; Kang, Y.-A.; Kang, H.; Woo, J.M.; Lee, Y.-S.; Jeon, M.; et al. Incidence, Management, and Prognosis of Graft Failure and Autologous Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. J. Korean Med. Sci. 2021, 36, e151. [Google Scholar] [CrossRef] [PubMed]
- Servais, S.; Beguin, Y.; Baron, F. Emerging drugs for prevention of graft failure after allogeneic hematopoietic stem cell transplantation. Expert Opin. Emerg. Drugs 2013, 18, 173–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astor, B.C.; Melamed, M.L.; Mandelbrot, D.A.; Djamali, A. Seasonality of mortality and graft failure among kidney transplant recipients in the US—A retrospective study. Transpl. Int. 2018, 31, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Tinckam, K. Histocompatibility methods. Transpl. Rev. 2009, 23, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Terasaki, P.I. Humoral theory of transplantation: Mechanism, prevention, and treatment. Hum. Immunol. 2005, 66, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Lentine, K.L.; Smith, J.; Hart, A.; Miller, J.; Skeans, M.; Larkin, L.; Robinson, A.; Gauntt, K.; Israni, A.; Hirose, R.; et al. OPTN/SRTR 2020 Annual Data Report: Kidney. Am. J. Transpl. 2022, 22 (Suppl. 2), 21–136. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Ghobrial, R.M. Chronic allograft rejection: A significant hurdle to transplant success. Burn. Trauma 2014, 2, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Bestard, O.; Nickel, P.; Cruzado, J.M.; Schoenemann, C.; Boenisch, O.; Sefrin, A.; Grinyó, J.M.; Volk, H.-D.; Reinke, P. Circulating Alloreactive T Cells Correlate with Graft Function in Longstanding Renal Transplant Recipients. J. Am. Soc. Nephrol. 2008, 19, 1419–1429. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, K.; Sandner, S.; Imitola, J.; Sho, M.; Li, Y.; Langmuir, P.B.; Rothstein, D.M.; Strom, T.B.; Turka, L.A.; Sayegh, M.H. Th1 cytokines, programmed cell death, and alloreactive T Cell clone size in transplant tolerance. J. Clin. Investig. 2002, 109, 1471–1479. [Google Scholar] [CrossRef]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 71, 189–194. [Google Scholar] [CrossRef]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T Cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhulai, G.; Oleinik, E. Targeting regulatory T cells in anti-PD-1/PD-L1 cancer immunotherapy. Scan. J. Immunol. 2022, 95, e13129. [Google Scholar]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T Cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [Green Version]
- Bretscher, P.A. A two-step, two-signal model for the primary activation of precursor helper T Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Duke-Cohan, J.S.; Chaudhri, A.; Aksoylar, H.-I.; Wang, Q.; Council, A.; Berg, A.; Freeman, G.J.; Boussiotis, V.A. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 2020, 3, 128. [Google Scholar] [CrossRef] [Green Version]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T Cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T Cells and its functional implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T Cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleman, L.J.; van Puijenbroek, A.A.F.L.; Shu, K.M.; Nadler, L.M.; Boussiotis, V.A. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T Cells. J. Immunol. 2002, 168, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.L. PD-1 signaling in primary T Cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol 2018, 103, 1065–1076. [Google Scholar] [CrossRef]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T Cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef]
- Deng, R.; Cassady, K.; Li, X.; Yao, S.; Zhang, M.; Racine, J.; Lin, J.; Chen, L.; Zeng, D. B7H1/CD80 interaction augments PD-1-dependent T Cell apoptosis and ameliorates graft-versus-host disease. J. Immunol. 2015, 194, 560–574. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.-M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T Cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Li, X.; Yao, S.; Wang, L.; Chen, Y.; Zhao, D.; Johnston, H.F.; Young, J.S.; Liu, H.; Todorov, I.; et al. Host APCs augment in vivo expansion of donor natural regulatory T Cells via B7H1/B7.1 in allogeneic recipients. J. Immunol. 2011, 186, 2739–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-J.; Omiya, R.; Matsumura, Y.; Sakoda, Y.; Kuramasu, A.; Augustine, M.M.; Yao, S.; Tsushima, F.; Narazaki, H.; Anand, S.; et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 2010, 116, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rollins, M.R.; Johnson, R.M.G. CD80 Expressed by CD8+ T Cells Contributes to PD-L1-Induced Apoptosis of Activated CD8+ T Cells. J. Immunol. Res. 2017, 2017, 7659462. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Ma, S.; Gong, H.; Wang, J.; Xu, Y.; Wu, D.; Sun, A. PD-L1 Ameliorates Murine Acute Graft-versus-Host Disease by Suppressing Effector but Not Regulatory T Cells Function. Arch. Immunol. Ther. Exp. 2019, 67, 179–187. [Google Scholar] [CrossRef]
- Blazar, B.R.; Carreno, B.M.; Panoskaltsis-Mortari, A.; Carter, L.; Iwai, Y.; Yagita, H.; Nishimura, H.; Taylor, P.A. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J. Immunol. 2003, 171, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Deng, R.; He, W.; Liu, C.; Wang, M.; Young, J.; Meng, Z.; Du, C.; Huang, W.; Chen, L.; et al. Loss of B7-H1 expression by recipient parenchymal cells leads to expansion of infiltrating donor CD8+ T Cells and persistence of graft-versus-host disease. J. Immunol. 2012, 188, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; O’connor, R.S.; Thangavelu, G.; Lovitch, S.; Dandamudi, D.B.; Wilson, C.B.; Vincent, B.G.; Tkachev, V.; Pawlicki, J.M.; Furlan, S.; et al. Programmed death ligand-1 expression on donor T Cells drives graft-versus-host disease lethality. J. Clin. Investig. 2016, 126, 2642–2660. [Google Scholar] [CrossRef]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.-Y.; Yuan, Y.; Li, Y.-C.; Yang, L.-Y.; Zhou, X.-Y.; Xiong, D.-Q.; Zhao, Z.-Y. Programmed Cell Death Ligand 1-Transfected Mouse Bone Marrow Mesenchymal Stem Cells as Targeted Therapy for Rheumatoid Arthritis. BioMed Res. Int. 2021, 2021, 5574282. [Google Scholar] [CrossRef]
- Sugiura, D.; Okazaki, I.-M.; Maeda, T.K.; Maruhashi, T.; Shimizu, K.; Arakaki, R.; Takemoto, T.; Ishimaru, N.; Okazaki, T. PD-1 agonism by anti-CD80 inhibits T Cell activation and alleviates autoimmunity. Nat. Immunol. 2022, 23, 399–410. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.E.; Liang, S.C.; Wu, Y.; Chernova, T.; Sobel, R.A.; Klemm, M.; Kuchroo, V.K.; Freeman, G.J.; Sharpe, A.H. PD-L1-deficient mice show that PD-L1 on T Cells, antigen-presenting cells, and host tissues negatively regulates T Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10691–10696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasagi, S.; Kawano, S.; Okazaki, T.; Honjo, T.; Morinobu, A.; Hatachi, S.; Shimatani, K.; Tanaka, Y.; Minato, N.; Kumagai, S. Anti-programmed cell death 1 antibody reduces CD4+PD-1+ T Cells and relieves the lupus-like nephritis of NZB/W F1 mice. J. Immunol. 2010, 184, 2337–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Albin, M.J.; Yuan, X.; Yamaura, K.; Habicht, A.; Murayama, T.; Grimm, M.; Waaga, A.M.; Ueno, T.; Padera, R.F.; et al. PDL1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J. Immunol. 2007, 179, 5204–5210. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Fujino, M.; Jiang, G.; Kitazawa, Y.; Xie, L.; Azuma, M.; Yagita, H.; Nagao, S.; Sugioka, A.; Kurosawa, Y.; et al. PD-1/B7-H1 interaction contribute to the spontaneous acceptance of mouse liver allograft. Am. J. Transpl. 2010, 10, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Tokita, D.; Mazariegos, G.V.; Zahorchak, A.F.; Chien, N.; Abe, M.; Raimondi, G.; Thomson, A.W. High PD-L1/CD86 ratio on plasmacytoid dendritic cells correlates with elevated T-regulatory cells in liver transplant tolerance. Transplantation 2008, 85, 369–377. [Google Scholar] [CrossRef]
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef]
- Hall, K.H.; Liu, Y.; Jiang, C.; Harvey, R.D. New and Worsening Long-term Immune-Related Adverse Events with PD-1/PD-L1 Pathway Agents in Patients with Cancer. Pharmacotherapy 2020, 40, 133–141. [Google Scholar] [CrossRef]
- Delanoy, N.; Michot, J.-M.; Comont, T.; Kramkimel, N.; Lazarovici, J.; Dupont, R.; Champiat, S.; Chahine, C.; Robert, C.; Herbaux, C.; et al. Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: A descriptive observational study. Lancet Haematol 2019, 6, e48–e57. [Google Scholar] [CrossRef] [PubMed]
- Canavan, M.; Floudas, A.; Veale, D.J.; Fearon, U. The PD-1:PD-L1 axis in Inflammatory Arthritis. BMC Rheumatol. 2021, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Jiang, L.; Li, S.-C.; He, Q.-J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Gao, Y.; Wei, W.; Zhang, J. Extracellular and nuclear PD-L1 in modulating cancer immunotherapy. Trends Cancer 2021, 7, 837–846. [Google Scholar] [CrossRef]
- Ying, H.; Zhang, X.; Duan, Y.; Lao, M.; Xu, J.; Yang, H.; Liang, T.; Bai, X. Non-cytomembrane PD-L1: An atypical target for cancer. Pharmacol. Res. 2021, 170, 105741. [Google Scholar] [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef]
- Elalouf, A. Infections after organ transplantation and immune response. Transpl. Immunol. 2023, 77, 101798. [Google Scholar] [CrossRef]
- Vietzen, H.; Furlano, P.L.; Cornelissen, J.J.; Böhmig, G.A.; Jaksch, P.; Puchhammer-Stöckl, E. HLA-E-restricted immune responses are crucial for the control of EBV infections and the prevention of PTLD. Blood 2023, 141, 1560–1573. [Google Scholar] [CrossRef]
- Park, J.-I.; Song, G.-W.; Ryu, J.H.; Choi, S.-T.; Choi, N.-G.; Jung, B.-H.; Chu, C.W.; Kim, K.-K.; Jung, D.-H.; Ha, T.-Y.; et al. A Multicenter, Randomized, Open-Label Study to Compare the Efficacy and Safety of Tacrolimus and Corticosteroids in Combination with or without Mycophenolate Mofetil in Liver Transplantation Recipients Infected with Hepatitis B Virus. Transplant Proc. 2023, 55, 387–395. [Google Scholar] [CrossRef]
- Burek Kamenaric, M.; Ivkovic, V.; Kovacevic Vojtusek, I.; Zunec, R. The Role of HLA and KIR Immunogenetics in BK Virus Infection after Kidney Transplantation. Viruses 2020, 12, 1417. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Handelsman, S.; Overbey, J.; Chen, K.; Lee, J.; Haj, D.; Li, Y. PD-L1’s Role in Preventing Alloreactive T Cell Responses Following Hematopoietic and Organ Transplant. Cells 2023, 12, 1609. https://doi.org/10.3390/cells12121609
Handelsman S, Overbey J, Chen K, Lee J, Haj D, Li Y. PD-L1’s Role in Preventing Alloreactive T Cell Responses Following Hematopoietic and Organ Transplant. Cells. 2023; 12(12):1609. https://doi.org/10.3390/cells12121609
Chicago/Turabian StyleHandelsman, Shane, Juliana Overbey, Kevin Chen, Justin Lee, Delour Haj, and Yong Li. 2023. "PD-L1’s Role in Preventing Alloreactive T Cell Responses Following Hematopoietic and Organ Transplant" Cells 12, no. 12: 1609. https://doi.org/10.3390/cells12121609